Agammaglobulinemia (مرض بروتون ، نقص سكر الدم الوراثي). Agammaglobulinemia Bruton. الأعراض والتشخيص والعلاج مرض الوحشية
- هذا مرض وراثي يتطور فيه نقص المناعة الأولي الشديد (خلل في الدفاع المناعي للجسم) مع انخفاض واضح في مستوى جاما جلوبيولين في الدم. يتجلى المرض عادة في الأشهر والسنوات الأولى من حياة الطفل ، عندما يتكرر الالتهابات البكتيرية: التهاب الأذن الوسطى ، التهاب الجيوب الأنفية ، الالتهاب الرئوي ، تقيح الجلد ، التهاب السحايا ، تعفن الدم. عندما يتم فحصها في الدم المحيطي ونخاع العظام ، فإن الغلوبولين المناعي في الدم والخلايا البائية غائبة عمليًا. علاج غاماغلوبولين الدم هو علاج بديل مدى الحياة.
التصنيف الدولي للأمراض - 10
D80نقص المناعة مع نقص الأجسام المضادة السائد

معلومات عامة
Agammaglobulinemia (نقص غاماغلوبولين الدم الوراثي ، مرض بروتون) هو عيب خلقي في المناعة الخلطية ناتج عن طفرات في جينوم الخلية ، مما يؤدي إلى عدم كفاية تخليق الخلايا اللمفاوية البائية. نتيجة لذلك ، يتم تعطيل تكوين الغلوبولين المناعي من جميع الفئات ، وينخفض محتواها في الدم بشكل حاد حتى يختفي تمامًا ، ويتطور نقص المناعة الأولية. يؤدي ضعف تفاعل الجهاز المناعي إلى تطور أمراض التهابية قيحية متكررة وخيمة في الجهاز التنفسي العلوي والشعب الهوائية والرئتين والجهاز الهضمي والسحايا. يحدث مرض بروتون حصريًا عند الأولاد ويحدث في حوالي 1-5 من بين مليون مولود جديد ، بغض النظر عن العرق أو المجموعة العرقية.
الأسباب
يحدث الشكل المرتبط بالكروموسوم X من agammaglobulinemia الوراثي بسبب تلف أحد جينات الكروموسوم X (الموجود في Xq21.3-22.2). هذا الجين مسؤول عن تخليق إنزيم التيروزين كيناز ، الذي يشارك في تكوين وتمايز الخلايا البائية. نتيجة للطفرات في هذا الجين وإعاقة تخليق التيروزين كيناز لبروتون ، تعطل تكوين المناعة الخلطية. في agammaglobulinemia ، توجد أشكال صغيرة (خلايا ما قبل B) في نخاع العظم ، ويضعف تمايزها ودخولها إلى مجرى الدم. وفقًا لذلك ، لا يتم إنتاج جميع فئات الغلوبولين المناعي عمليًا ، ويصبح جسم الطفل أعزل عندما تخترق البكتيريا المسببة للأمراض (غالبًا ما تكون هذه المكورات العقدية والمكورات العنقودية والزائفة الزنجارية).
لوحظ وجود آلية مماثلة للاضطرابات في حالة وجود شكل آخر من أشكال agammaglobulinemia الوراثي - المرتبط بالكروموسوم X ونقص هرمون النمو. يتطور الشكل المتنحي الجسدي نتيجة للطفرات في العديد من الجينات (سلاسل µ الثقيلة ، وجين λ5 / 14.1 ، وجين البروتين المحول ، وجين جزيء إشارة IgA).
تصنيف
هناك ثلاثة أشكال وراثية لداء الغاماغلوبولين في الدم:
- مرتبط بالكروموسوم X (85٪ من جميع حالات نقص السكر في الدم الخلقي ، يتأثر الأولاد فقط)
- نوع سويسري متنحي متنحي متقطع (يحدث عند الأولاد والبنات)
- agammaglobulinemia مرتبط بالكروموسوم X ونقص هرمون النمو (نادر جدًا وفقط عند الأولاد)
أعراض الإصابة بغاماغلوبولين الدم
يؤدي انخفاض تفاعل المناعة الخلطية في الإصابة بضادات الغاما غلوبولين الدم إلى تطور أمراض التهابية قيحية متكررة بالفعل في السنة الأولى من حياة الطفل (عادةً بعد ذلك الرضاعة الطبيعية- في عمر 6-8 أشهر). في الوقت نفسه ، لم تعد الأجسام المضادة الواقية من الأم تدخل جسم الطفل ، ولا يتم إنتاج الغلوبولين المناعي الخاص به. بحلول سن 3-4 سنوات ، تصبح العمليات الالتهابية مزمنة مع ميل إلى التعميم. يمكن أن تؤثر العدوى القيحية مع agammaglobulinemia على أعضاء وأنظمة مختلفة.
من جانب أعضاء الأنف والأذن والحنجرة ، فإن التهاب الجيوب الأنفية القيحي ، والتهاب الإيثويد ، والتهاب الأذن الوسطى ليس من غير المألوف ، وغالبًا ما يتطور التهاب الأذن الوسطى القيحي في السنة الأولى من حياة الطفل ، والتهاب الجيوب الأنفية - في 3-5 سنوات. من أمراض الجهاز القصبي الرئوي ، لوحظ التهاب الشعب الهوائية المتكرر والالتهاب الرئوي وخراجات الرئة.
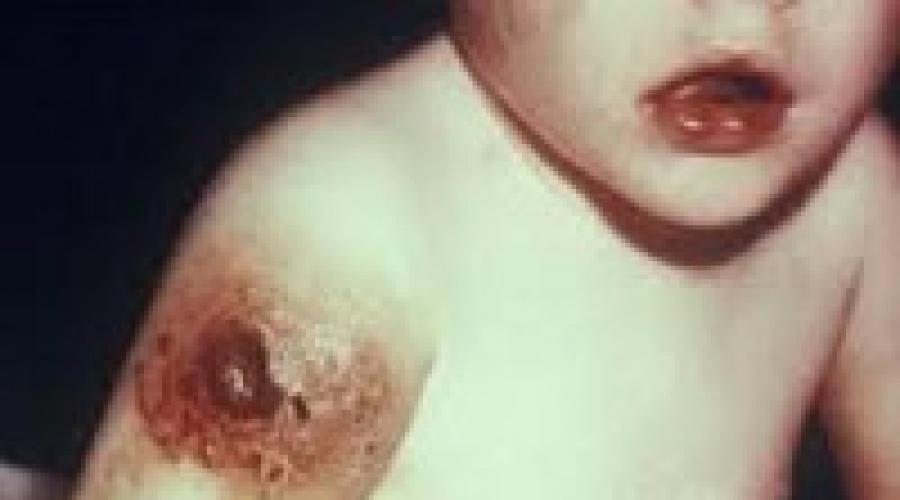
غالبًا ما تكون هناك آفة في الجهاز الهضمي مع الإسهال المستمر (الإسهال) الناجم عن التهاب الأمعاء والقولون المعدي المزمن (مسببات الأمراض الرئيسية هي العطيفة والجيارديا والفيروس العجلي). على ال جلدتم العثور على القوباء ، والأكزيما الجرثومية ، وداء الدمامل المتكرر ، والخراجات والفلغمون.
ليس من غير المألوف أن تتلف العينين (التهاب الملتحمة القيحي) ، وتجويف الفم (التهاب الفم التقرحي ، والتهاب اللثة) ، والجهاز العضلي الهيكلي (التهاب العظم والنقي ، والتهاب المفاصل القيحي). بشكل عام ، تتميز الصورة السريرية لداء غاماغلوبولين الدم بمجموعة من الأعراض العامة التي لوحظت مع عدوى قيحية (ارتفاع درجة حرارة الجسم ، قشعريرة ، آلام عضلية وصداع ، ضعف عام ، اضطراب في النوم والشهية ، إلخ) وعلامات تلف في الجسم. عضو معين (سعال ، ضيق في التنفس ، صعوبة في التنفس عن طريق الأنف ، إفراز صديدي ، إسهال ، إلخ).
المضاعفات
أي مرض معدي وجسمي في مريض نقص المناعة يكون شديدًا وطويل الأمد ويصاحبه مضاعفات. يمكن أن يكون المسار الحاد لعدوى غاماغلوبولين الدم معقدًا بسبب تطور التهاب السحايا والتهاب الدماغ والنخاع الفيروسي وشلل الأطفال بعد التطعيم والإنتان. على خلفية المرض ، تزداد احتمالية الإصابة بأمراض المناعة الذاتية والأورام. غالبًا ما تحدث وفاة المرضى بسبب الصدمة السامة المعدية.
التشخيص
أثناء الفحص السريري للمريض من قبل أخصائي أمراض الحساسية والمناعة ، يتم الكشف عن علامات الآفة التهابية قيحية لعضو معين (نسيج) وأعراض تؤكد انخفاض تفاعل الجهاز المناعي: نقص تنسج اللوزتين ، انخفاض في الغدد الليمفاوية المحيطية. كما يتم التعبير عن علامات تأخر في النمو البدني للطفل.
يكشف اختبار الدم المخبري عن انخفاض واضح في مستوى الغلوبولين المناعي في جهاز المناعة (IgA و IgM< 20 мг/дл – снижение в сто раз, IgG < 200 мг/дл – в десять раз) или их полное отсутствие. В периферической крови обнаруживается глубокий дефицит B-клеток (менее 1%), в костном мозге практически отсутствуют плазмоциты. Что касается клеточного иммунитета, то содержание T-лимфоцитов может быть в норме. При гистологическом исследовании лимфоидной ткани герминативные (зародышевые) центры и плазматические клетки отсутствуют.
يتم إجراء التشخيص التفريقي للعدوى غاماغلوبولين الدم الوراثي مع حالات نقص المناعة الأولية والثانوية الأخرى (الاضطرابات الوراثية ، عدوى فيروس نقص المناعة البشرية والفيروس المضخم للخلايا ، الحصبة الألمانية الخلقية وداء المقوسات ، الأورام الخبيثة والاضطرابات الجهازية ، نقص المناعة بسبب تسمم الأدوية ، إلخ).
علاج مرض غاماغلوبولين الدم
مطلوب علاج بديل مدى الحياة بالأدوية المحتوية على الأجسام المضادة. عادة ، يتم استخدام الغلوبولين المناعي في الوريد ، وفي غيابه ، يتم استخدام البلازما الأصلية من متبرعين أصحاء دائمين. عندما يتم تشخيص agammaglobulinemia لأول مرة ، يتم إجراء العلاج البديل في وضع التشبع حتى يصل مستوى الغلوبولين المناعي IgG إلى أكثر من 400 مجم / ديسيلتر ، وبعد ذلك ، في حالة عدم وجود عملية التهابات قيحية نشطة في الأعضاء والأنسجة يمكن المضي قدمًا في علاج الصيانة بإدخال الجرعات الوقائية من الأدوية التي تحتوي على الغلوبولين المناعي.
أي حلقة من عدوى قيحية بكتيرية ، بغض النظر عن مكان العملية الالتهابية ، تتطلب كمية كافية العلاج بالمضادات الحيوية، والتي يتم إجراؤها في وقت واحد مع العلاج البديل. في كثير من الأحيان ، مع agammaglobulinemia ، يتم استخدام العوامل المضادة للبكتيريا من مجموعة السيفالوسبورينات ، والأمينوغليكوزيدات ، والماكروليدات ، وكذلك المضادات الحيوية للبنسلين. مدة العلاج في هذه الحالة أعلى بعدة مرات من المعيار لهذا المرض.
يتم إجراء علاج الأعراض مع مراعاة الآفة المحددة لعضو معين (غسل الجيوب الأنفية بالمطهرات وإجراء تدليك بالاهتزاز صدروالصرف الوضعي في التهاب الشعب الهوائية والالتهاب الرئوي ، وما إلى ذلك).
تنبؤ بالمناخ
إذا تم الكشف عن agammaglobulinemia في سن مبكرة قبل ظهور المضاعفات الشديدة ، وبدء العلاج البديل المناسب لحالة المريض في الوقت المناسب ، فمن الممكن الحفاظ على نمط حياة طبيعي لسنوات عديدة. ومع ذلك ، في معظم الحالات ، يتم تشخيص الاضطرابات الوراثية للمناعة الخلطية بعد فوات الأوان ، عندما تتطور بالفعل أمراض التهابات قيحية مزمنة لا رجعة فيها لأعضاء وأنظمة الجسم. في هذه الحالة ، يكون تشخيص الإصابة بغاماغلوبولين الدم غير موات.
مرض بروتون (agammaglobulinemia الخلقي ، agammaglobulinemia المرتبط بـ X) هو نقص مناعي خلطي أساسي ، والذي ينتج عن التغيرات الطفرية في الجين المشفر غير المستقبل للتيروزين كيناز.
| التصنيف الدولي للأمراض - 10 | D80.0 |
|---|---|
| التصنيف الدولي للأمراض - 9 | 279.04 |
| الأمراض | 1728 |
| OMIM | 300300 |
| إي ميديسين | بيد / 294 ديرم / 858 |
معلومات عامة
تم وصف agammaglobulinemia المرتبط بـ X لأول مرة في عام 1952 من الولايات المتحدة الأمريكية بواسطة Ogden Bruton. لاحظ الطبيب وجود طفل ، من سن 4 سنوات ، كان مريضًا بمختلف الأمراض المعدية الوخيمة أكثر من عشر مرات ، ولم يتم العثور على أجسام مضادة في دمه. تم إنشاء الطبيعة الجينية لمرض بروتون في عام 1993.
يؤثر Agammaglobulinemia على الذكور فقط. يتم تشخيص المرض في 1 من كل 250.000 فتى.
الأسباب
سبب مرض بروتون هو وجود بروتين متحور في الجين يشفر كيناز التيروزين غير المستقبلي (بروتون تيروزين كيناز أو TKB). يقع هذا الجين على إحدى قواعد الكروموسوم X ، لذلك يُطلق على علم الأمراض اسم مرتبط بالكروموسوم X.
المرض موروث بطريقة متنحية. يظهر فقط في الأولاد ، لأن الحمض النووي الخاص بهم يحتوي على كروموسوم X واحد وكروموسوم Y واحد. البنات لديهن اثنان من الكروموسومات X ، لذلك يتم تعويض الجين المعيب بالكروموسوم الطبيعي. لكن النساء يحملن الجين المعدل وينقلنه إلى أبنائهن.
في حالات نادرة ، يكون agammaglobulinemia في بروتون غير وراثي: التغييرات في الجين المشفر TKB تحدث مباشرة بعد الحمل.
طريقة تطور المرض
إن جين التيروزين كيناز غير المستقبل هو المسؤول عن نضوج الخلايا الليمفاوية البائية ، وهي الخلايا التي تلعب دورًا مهمًا في المناعة الخلطية. عند التلامس مع مستضد (فيروس أو بكتيريا) ، يتحول بعضها إلى خلايا بلازما تنتج أجسامًا مضادة (الغلوبولين المناعي) ، بينما يتحول البعض الآخر إلى خلايا ذاكرة ب.
يشير agammaglobulinemia إلى هزيمة المناعة الخلطية. بسبب طفرة الجين TKB ، يتم حظر نضج الخلايا الليمفاوية B ، ونتيجة لذلك يفقد الجسم القدرة على إنتاج الغلوبولين المناعي بشكل كامل عند ملامسته لعامل معدي. يمكن أن تختلف شدة التغيرات المرضية من انخفاض كبير في مستوى الأجسام المضادة في الدم إلى غيابها التام.
كقاعدة عامة ، المرضى الذين يعانون من agammaglobulinemia المرتبط بـ X في الدم المحيطي يظهرون القليل من الخلايا الليمفاوية B أو لا يظهرون على الإطلاق وتركيز منخفض لجميع فئات الغلوبولين المناعي ، بالإضافة إلى عدم وجود خلايا البلازما في الأنسجة اللمفاوية. يؤدي التوليف غير الكافي للأجسام المضادة إلى حقيقة أن الجسم لا يستطيع التعامل مع العوامل المعدية ، وخاصة البكتيريا.
يتميز مرض بروتون باستمرار الاستجابة المناعية له اصابات فيروسية. في هذا يختلف عن علم الأمراض الوراثي يسمى agammaglobulinemia من النوع السويسري ، والذي يتميز بخلل في المناعة الخلوية والخلطية. يعتمد تطوره على نقص أو عدم وجود الخلايا الليمفاوية B و T ، مما يجعل الشخص عرضة للعدوى من أي مسببات. يصيب المرض كلا الجنسين.
أعراض
تبدأ أعراض مرض بروتون بالظهور من 4-6 أشهر ، حيث ينخفض عدد الأجسام المضادة المنقولة من الأم في دم الطفل. يتمثل العرض الرئيسي لعدوى غاماغلوبولين الدم مع نقص الخلايا البائية في الالتهابات المزمنة والمتكررة التي تسببها البكتيريا القيحية ، أي الكائنات الحية الدقيقة التي يمكن أن تسبب التهابًا صديديًا. وتشمل هذه المكورات الرئوية والمكورات العنقودية والمستدمية النزلية وغيرها.
يعاني الطفل من أمراض الجهاز التنفسي العلوي والجهاز التنفسي والجهاز الهضمي والجلد والدهون تحت الجلد وما إلى ذلك. أكثر الأمراض شيوعًا هي الالتهاب الرئوي والتهاب الأذن والتهاب الجيوب والتهاب الملتحمة والأكزيما والتهاب الجلد والعضلات والتهاب السحايا والتهاب الدماغ.
تستمر مقاومة الفيروسات في مرض بروتون ولكنها تضعف ، مما يتسبب في حدوث مضاعفات. يؤدي فيروس التهاب الكبد B إلى التهاب الكبد التدريجي ، والفيروسة العجلية - إلى متلازمة سوء الامتصاص والإسهال المزمن ، والتلامس مع فيروس شلل الأطفال أثناء التطعيم - إلى شلل الأطفال.
الأطفال المصابون بمرض بروتون معرضون لردود الفعل التحسسية وأمراض المناعة الذاتية وأمراض الأورام وأمراض النسيج الضام (التهاب المفاصل الكبيرة).
المظاهر الأخرى لداء غاماغلوبولين الدم:
- عدم الاستجابة من الجهاز اللمفاويفي فترة حادةالأمراض.
- صغر حجم اللوزتين.
- توسع القصبات هو توسع القصبات ، مصحوبا بنوبات الربو.
في الأطفال الذين يعانون من خلل في المناعة الخلطية ، لا يوجد تضخم في اللوزتين الأنفية والبلعومية.
التشخيص
يتم تشخيص مرض بروتون على أساس سوابق المريض وفحص المريض والطرق الفعالة والاختبارات المعملية. يشار إلى علم الأمراض من خلال الأمراض البكتيرية المتكررة في التاريخ. تشمل العلامات الأخرى لعدوى غاماغلوبولين الدم التي يمكن اكتشافها باستخدام الأشعة السينية التخلف (غياب) اللوزتين والعقد الليمفاوية ، وكذلك التغيرات في بنية الطحال.
معظم طريقة إعلاميةتشخيص مرض بروتون - فحص الدم ، وعلى وجه الخصوص:
- قياس التدفق الخلوي - يُظهر عددًا أقل من الخلايا الليمفاوية ب أو غيابها ؛
- الرحلان الكهربي المناعي في الدم - يدل على عدم وجود جزء صغير من جاما الجلوبيولين ؛
- قياس الكلى - يسمح لك باكتشاف تركيز غير كافٍ من الغلوبولين المناعي (يتم تقليل مستوى Ig A و Ig M بمقدار 100 مرة ، Ig G - بمقدار 10 مرات) ؛
- التحليل العام - يوضح انحرافًا عن القاعدة في عدد الكريات البيض.
بالإضافة إلى ذلك ، يتم إجراء دراسة وراثية جزيئية ، يتم خلالها اكتشاف خلل في الجين المشفر غير المستقبل للتيروزين كيناز. يمكن إجراء هذا الاختبار في مرحلة التخطيط أو أثناء الحمل.
علاج او معاملة
تتمثل المبادئ الرئيسية لعلاج غاماغلوبولين الدم في الحفاظ على الجهاز المناعي طوال الحياة والعلاج بالمضادات الحيوية في تطوير الأمراض المعدية.
للتعويض عن نقص المناعة ، يتم إجراء العلاج البديل بمستحضرات جاما الجلوبيولين. يتم إعطاء الأدوية عن طريق الوريد بمعدل 400 مجم لكل 1 كجم من وزن الجسم. الهدف من العلاج الدوائي هو تحقيق تركيز من الغلوبولين المناعي في الدم يساوي 3 جم / لتر.
في الفترة الحادة أمراض معديةمع agammaglobulinemia Bruton ، يتم استخدام المضادات الحيوية: السيفالوسبورينات ، السلفوناميدات ، البنسلين ، الأمينوغليكوزيدات.
تنبؤ بالمناخ
مرض بروتون له تشخيص إيجابي مع استمرار إعطاء الغلوبولين المناعي والعلاج المناسب بالمضادات الحيوية. التطبيق في الوقت المناسب العوامل المضادة للبكتيريامع تفاقم الأمراض المعدية ، يمكن أن يؤدي إلى التطور السريع للعملية المرضية والوفاة.
الوقاية
نظرًا لأن غاماغلوبولين الدم في بروتون وراثي بطبيعته ، فإن الوقاية منه غير ممكنة. يُنصح بإجراء استشارة وراثية عند التخطيط للحمل للأزواج الذين لديهم تاريخ عائلي لهذا المرض.
إذا كان الطفل مصابًا بغاماغلوبولين الدم ، فيجب اتخاذ تدابير لمنع حدوث مضاعفات وتكرار العدوى. وتشمل هذه:
- إعادة تأهيل بؤر الالتهاب المزمنة.
- العلاج المناسب للأمراض.
- التطعيم بالأدوية المعطلة فقط.
النسخة المطبوعة
إرسال عملك الجيد في قاعدة المعرفة أمر بسيط. استخدم النموذج أدناه
سيكون الطلاب وطلاب الدراسات العليا والعلماء الشباب الذين يستخدمون قاعدة المعرفة في دراساتهم وعملهم ممتنين جدًا لك.
نشر على http://www.allbest.ru/
نشر على http://www.allbest.ru/
ملخص عن الموضوع:
"مرض ومتلازمة بروتون"
المقدمة
1. شروط نقص المناعة
2. نقص المناعة الأولية
4. داء بروتون
4.1 الأسباب
4.2 الفيزيولوجيا المرضية
4.3 الأعراض والمظاهر
4.4 التشخيص
4.5 العلاج
4.6 المضاعفات
4.7 الوقاية
استنتاج
فهرس
المقدمة
حالات نقص المناعة (IDS) - تغييرات مستمرة أو مؤقتة حالة المناعةناتج عن خلل في آلية أو أكثر من آليات الاستجابة المناعية للتعرض لمستضد.
1. شروط نقص المناعة
تصنيف حالات نقص المناعة (IDS)
I. حسب الأصل ، يتم تصنيف IDS على النحو التالي:
1) أساسي (وراثي)
2) الثانوية (المكتسبة)
أ) فسيولوجية
ب) المرضية
II. وفقًا لآليات التطوير ، تتميز مجموعات IDS التالية:
الآلية الأولى مستحقة
1) غياب أو نقص عدد الخلايا المساعدة (ASCs) ، أي الخلايا وحيدة النواة - الضامة.
2) غياب أو نقص عدد الخلايا الليمفاوية B ؛
3) غياب أو نقص عدد الخلايا اللمفاوية التائية ومجموعاتها السكانية الفرعية في نظام T ؛
4) غياب أو نقص عدد الخلايا لجميع الفئات المدرجة في ICS ، أي أشكال مجتمعة من IDS ؛
5) غياب أو تقليل الخلايا السليفة ICS بسبب الحصار المفروض على نضجها أو تدميرها.
ترتبط الآلية الثانية بالاضطرابات في عمليات تنظيم تمايز الخلايا في النظامين B و T ، بالإضافة إلى تعاونهما والخلايا الأخرى في تنفيذ الاستجابة المناعية.
ثالثا. وفقًا للضرر السائد لخلايا أنظمة ICS المختلفة ، يتم تمييز ما يلي:
1) IDS المعتمدة على B أو الخلطية ؛
2) IDS المعتمدة على T أو الخلوية ؛
3) البلعمة IDS ("A المعتمد") ؛
4) IDS مجتمعة - تلف الآليات الخلوية والخلطية للمناعة (على سبيل المثال ، الخلايا الليمفاوية B و T).
رابعا. قد تترافق أعراض ضعف المناعة مع:
مع عدم وجود عدد غير كافٍ و / أو وظيفة محدودة لخلايا ICS ، فضلاً عن ضعف البلعمة والمكونات التكميلية. يتم مواجهة أشكال مختلفة من IDS بشكل مختلف - غالبًا ما تتضرر آليات المناعة الخلطية ، وغالبًا ما يتم العثور على انتهاكات للأشكال الخلوية والمجمعة من المناعة ؛ الاضطرابات الأخرى ، تسمى حالات نقص المناعة غير النوعي (عيوب في النظام التكميلي والبلعمة) ، نادرة للغاية:
الاضطرابات الخلطية 75٪
أشكال مجمعة من IDS 10-25٪
عيوب المناعة الخلوية 5-10٪
خلل في البلعمة 1-2٪
عيب البروتين التكميلي
2. نقص المناعة الأولية
تتجلى أوجه القصور المناعي الأولية (العيوب الوراثية والخلقية في الجهاز المناعي) في تطور الآفات المعدية في الجسم بعد الولادة بفترة وجيزة ، ولكن قد لا يكون لها مظاهر سريرية إلا في وقت لاحق من الحياة.
العيوب الجينية والكروموسومية (العديد من العيوب المناعية من فئات مختلفة).
حالات نقص المناعة الثانوية ، أو حالات نقص المناعة - يتطور نقص المناعة بسبب التأثيرات الداخلية والخارجية على الجهاز المناعي الطبيعي (على سبيل المثال ، حوالي 90 ٪ من جميع الالتهابات الفيروسية مصحوبة بتثبيط مناعي عابر).
تتنوع أسباب حالات نقص المناعة ، فهي تشمل:
الأدوية المثبطة للمناعة (بما في ذلك الفينيتوين ، البنسيلامين ، القشرانيات السكرية).
سوء التغذية وهضم التجويف والغشاء وامتصاص الأمعاء.
الأدوية والمواد السامة.
التعرض للإشعاع ، أدوية العلاج الكيميائي.
نمو الأورام الخبيثة.
الفيروسات (على سبيل المثال ، فيروس نقص المناعة البشرية).
الحالات التي تؤدي إلى فقدان البروتين (مثل المتلازمة الكلوية).
نقص الأكسجة.
قصور الغدة الدرقية.
اسبلينيا.
نقص المناعة الأولية
وفقًا لتسميات منظمة الصحة العالمية ، يُفهم النقص المناعي المنشأ الأساسي عمومًا على أنه عجز محدد وراثيًا عن الجسم لإدراك ارتباط واحد أو آخر من الاستجابة المناعية.
وفقًا للتصنيف الذي اقترحته منظمة الصحة العالمية ، اعتمادًا على الآفة السائدة للروابط B و T للجهاز المناعي ، يتم تمييز حالات نقص المناعة الأولية المحددة التالية:
مجتمعة ، مع نفس الدرجة من الشدة أو مختلفة ، الضرر الذي يلحق بالأجزاء الخلوية (T) والخلطية (B) من الجهاز المناعي ؛
مع تلف سائد للوصلة الخلوية (T) لجهاز المناعة ؛
مع تلف سائد في الارتباط الخلطي (ب) لجهاز المناعة (علم أمراض إنتاج الأجسام المضادة).
حالات نقص المناعة الأولية ، كقاعدة عامة ، نادرة. حسب التصنيف الدولي للأمراض ، هناك عدة أصناف متميزة.
يتميز العوز المناعي الشديد المركب T و B بتطور خلل في الهياكل ذات الكفاءة المناعية في المراحل الأولى من تطور الكائن الحي. سريريا هو الأصعب. يمكن أن يحدث موت الكائن الحي في الرحم ، في الأيام الأولى بعد الولادة بسبب الغياب أو التثبيط الحاد للخلايا الجذعية المكونة للدم ، وكذلك بسبب الغياب أو التثبيط الحاد للغدة الصعترية وأعضاء الجهاز المناعي الأخرى ، مع انخفاض متزامن وواضح في عدد الخلايا اللمفاوية التائية والخلايا البائية وخلايا البلازما. يتجلى سريريًا من خلال انخفاض حاد في تفاعل ومقاومة الجسم لتأثير العوامل المسببة للأمراض المختلفة ، بما في ذلك الفيروسات والبكتيريا والفطريات.
تحدث المتغيرات في حالات نقص المناعة المشترك بسبب عيوب وراثية تؤثر على خطوط مختلفة من تمايز الخلايا الليمفاوية ، وكذلك المراحل المبكرة من تطورها ، وهي مشتركة بين السكان T- و B.
مبادئ علاج IDS الأولي
يعتمد العلاج على نوع النقص المناعي الأولي ويشمل العلاج البديل المستهدف (زرع الأنسجة ذات الكفاءة المناعية ، وزرع الغدة الصعترية الجنينية ، ونخاع العظام ، وإعطاء الغلوبولين المناعي الجاهز - الغلوبولين ، والأجسام المضادة المركزة ، ونقل الدم المباشر من متبرعين محصنين ، وإدارة الغدة الصعترية الهرمونات).
يتم استخدام التحصين الفعال ضد العدوى المتكررة بمساعدة اللقاحات المقتولة ، ويتم إعطاء السلفوناميدات.
3. الثانوية (المكتسبة) دفاع المناعة
يعد نقص المناعة الثانوي أو المكتسب انتهاكًا للدفاع المناعي للجسم ، ويحدث في فترة ما بعد الولادة نتيجة لعمل عوامل خارجية أو داخلية ، غير مرتبطة بآفة أولية للجهاز الوراثي.
تعتبر حالات نقص المناعة الثانوية شائعة جدًا.
قائمة الأمراض الرئيسية المصحوبة بنقص المناعة الثانوي التي اقترحها خبراء منظمة الصحة العالمية.
1. الأمراض المعدية:
أ) أمراض البروتوزوا والديدان الطفيلية - الملاريا وداء المقوسات وداء الليشمانيات والبلهارسيا ، إلخ ؛
ب) الالتهابات البكتيرية - الجذام والسل والزهري والمكورات الرئوية والتهابات المكورات السحائية ؛
ج) الالتهابات الفيروسية - الحصبة ، والحصبة الألمانية ، والأنفلونزا ، والنكاف ، وجدري الماء ، والتهاب الكبد الحاد والمزمن ، وما إلى ذلك ؛
ز) الالتهابات الفطرية- داء المبيضات ، داء الكروانيديا ، إلخ.
2. الاضطرابات التغذوية - سوء التغذية ، والدنف ، واضطرابات الامتصاص المعوي ، إلخ.
3. التسمم الخارجي والداخلي - مع القصور الكلوي والكبدي ، والتسمم بمبيدات الأعشاب ، وما إلى ذلك.
4. أورام الأنسجة اللمفاوية (سرطان الغدد الليمفاوية ، ورم الغدة الصعترية ، ورم الغدد الليمفاوية) ، الأورام الخبيثةأي توطين.
5. أمراض التمثيل الغذائي (داء السكري ، وما إلى ذلك).
6. فقدان البروتين في أمراض الأمعاء ، المتلازمة الكلوية ، أمراض الحروق ، إلخ.
7. عمل مختلف أنواع الإشعاع وخاصة الإشعاعات المؤينة.
8. تأثيرات إجهاد قوية وطويلة الأمد.
9. العمل أدوية(مثبطات المناعة ، الكورتيكوستيرويدات ، المضادات الحيوية ، السلفوناميدات ، الساليسيلات ، إلخ).
10. الحصار بالمجمعات المناعية والأجسام المضادة للخلايا الليمفاوية في بعض أمراض الحساسية وأمراض المناعة الذاتية.
يمكن تقسيم CIDs الثانوية إلى شكلين رئيسيين:
1) جهازية ، تتطور نتيجة الضرر الجهازي لتكوين المناعة (مع آفات الإشعاع ، السامة ، المعدية ، الإجهاد) ؛
2) موضعي ، يتميز بضرر ناحي للخلايا المؤهلة مناعياً (اضطرابات موضعية في الجهاز المناعي للغشاء المخاطي والجلد والأنسجة الأخرى ، تتطور نتيجة للاضطرابات الالتهابية والضمورية ونقص الأكسجة الموضعية).
يرتبط نقص المناعة الفسيولوجية بأسباب محددة.
يتميز قصور الجهاز المناعي لحديثي الولادة بتدني الروابط الخلوية والخلطية ، فضلاً عن عوامل المقاومة غير النوعية.
يتم الجمع بين عدد كبير من الخلايا الليمفاوية في الدم المحيطي لحديثي الولادة مع انخفاض في النشاط الوظيفي للخلايا اللمفاوية التائية والبائية. يحدث تخليق الأجسام المضادة للمستضد بشكل أساسي بسبب IgM ، وينخفض محتوى IgG و IgA ويصل إلى مستويات البالغين فقط في سن 11-14. هناك نشاط بلعمي منخفض وقدرة طاردة للدم. يتم تقليل المستوى التكميلي ويعود إلى طبيعته بحلول الشهر الثالث والسادس من العمر.
تتميز الحالة المناعية للمرأة الحامل بانخفاض عدد ووظائف الخلايا الليمفاوية التائية والبائية ، والتي يمكن تفسيرها من خلال زيادة محتوى ونشاط مثبطات T. هذا ضروري لقمع الاستجابة المناعية للمضادات الخلقية الجنينية.
يتجلى عدم كفاية جهاز المناعة أثناء الشيخوخة في انخفاض نشاط الروابط الخلطية والخلوية. تنخفض مستويات الأجسام المضادة الطبيعية في الدم ، وتقل القدرة على تخليق الأجسام المضادة لتحفيز المستضد. هذه هي بشكل رئيسي أجسام مضادة منخفضة IgM. إنتاج الأجسام المضادة IgGو IgA يتم تقليله بشكل كبير.
تخليق الأجسام المضادة لـ lgE يُثبط ، وبالتالي فإن شدتها ردود الفعل التحسسيةيلين. يعاني الارتباط الخلوي للجهاز المناعي بشكل كبير. ينخفض العدد الإجمالي للخلايا الليمفاوية في الدم المحيطي ، وكذلك العدد النسبي والمطلق للخلايا اللمفاوية التائية والبائية ونشاطها الوظيفي. يتم تقليل النشاط البلعمي للخلايا الضامة والخلايا المحببة العدلة ونشاط المكمل والليزوز ونشاط مبيد الجراثيم في مصل الدم. تصبح تفاعلات المناعة الذاتية أكثر تكرارا وتصبح أكثر حدة مع تقدم العمر.
تتميز حالات نقص المناعة المرضية ، وكذلك الأولية (الوراثية) ، بزيادة وتيرة (عشرات وحتى مئات المرات) من الأورام الخبيثة (ساركومة شبكية ، ساركومة ليمفاوية ، إلخ) ، وخاصة سرطان الدم والأورام اللمفاوية. مع نقص المناعة الثانوي ، كما هو الحال مع حالات نقص المناعة الأولية ، قد تعاني المناعة الخلطية أو الخلوية أيضًا. وبالتالي ، فإن الأمراض المصحوبة بفقدان البروتينات غالبًا ما تؤدي إلى الإصابة بنقص المناعة الثانوي الخلطي: الحروق ، المتلازمة الكلوية ، التهاب الكلية المزمن ، إلخ.
تؤدي العدوى الفيروسية الشديدة (الحصبة والإنفلونزا) إلى تطور نقص المناعة الخلوي الثانوي. أمراض فطرية(داء المبيضات الخارجي والداخلي).
يمكن أن يحدث تطور نقص المناعة أيضًا من خلال خطأ الأطباء الذين استخدموا منذ فترة طويلة زراعة الأعضاء وعلاج العديد من الأمراض الخطيرة ، وخاصة الورم ، وأمراض المناعة الذاتية (التهاب المفاصل الروماتويدي) ، ومثبطات المناعة الالتهابية:
مستحضرات هرمونية كورتيكوستيرويد.
مثبطات تخليق البروتين.
مضادات حيوية؛
مضادات الأورام التثبيط. مضادات الأيض البيورين وبيريميدين ؛
التعرض للأشعة السينية ، إلخ.
مبادئ علاج IDS الثانوي
1. العلاج البديل - استخدام مستحضرات المناعة المختلفة (مستحضرات الجلوبيولين ، مضادات السموم ، مضادات الأنفلونزا ، الأمصال المضادة للمكورات العنقودية ، إلخ).
2. تصحيح ارتباط المستجيب. يشمل التأثيرات على جهاز المناعة المستحضرات الدوائيةالتي تصحح عملها (ديكاريس ، ديوسيفون ، إيموران ، سيكلوفوسفاميد ، إلخ) ، هرمونات ووسطاء جهاز المناعة (مستحضرات الغدة الصعترية - ثيموسين ، ثيمالين ، تي أكتيفين ، إنترفيرون كريات الدم البيضاء).
3. إزالة العوامل المثبطة التي تربط الأجسام المضادة وتمنع تأثير التصحيح المناعي (امتصاص الدم ، فصادة البلازما ، غسيل الكلى ، الفصادة الليمفاوية ، إلخ).
أصبحت متلازمة نقص المناعة المكتسبة (الإيدز) ذات أهمية متزايدة بين حالات نقص المناعة الثانوية في العقد الماضي. تم وصف الأخير لأول مرة في الأدبيات العلمية في عام 1981 من قبل العلماء الأمريكيين.
وفقًا لمنظمة الصحة العالمية ، في السنوات الأخيرة ، تضاعف عدد الأشخاص الذين يعانون من حالات الإيدز المسجلة كل ستة أشهر.
إذا كان عدد المصابين بفيروس نقص المناعة البشرية في وقت سابق يزيد 50-100 مرة عن عدد الحالات ، فقد بلغ عدد المصابين بالفعل في عام 2001 130 مليونًا ، بما في ذلك 35 مليون شخص الاعراض المتلازمةالمعينات.
4. داء بروتون
أو agammaglobulinemia Bruton ، هو نقص مناعي وراثي ناتج عن طفرات في الجين الذي يشفر بروتون التيروزين كيناز. تم وصف المرض لأول مرة بواسطة Bruton في عام 1952 ، وبعد ذلك تم تسمية الجين المعيب. إن كينازات التيروزين لبروتون مهمة في نضوج الخلايا البائية السابقة لتمايز الخلايا البائية الناضجة. تم العثور على جين Bruton tyrosine kinase على الذراع الطويلة للكروموسوم X في النطاق من Xq21.3 إلى Xq22 ؛ يتكون من 37.5 كيلو قاعدة مع 19 exons التي تشفر 659 من الأحماض الأمينية ؛ هذه الأحماض الأمينية هي التي تكمل تكوين العصارة الخلوية التيروزين كيناز. في هذا الجين ، تم بالفعل تسجيل 341 حدثًا جزيئيًا فريدًا. بالإضافة إلى الطفرات ، تم العثور على عدد كبير من المتغيرات أو الأشكال المتعددة.
متلازمة نقص المناعة المعدي
4.1 الأسباب
تتداخل الطفرات في الجين الذي يكمن وراء مرض بروتون مع تطور ووظيفة الخلايا الليمفاوية البائية ونسلها. الفكرة الرئيسية هي أن ملف الشخص السليمتنضج خلايا ما قبل B لتصبح خلايا ليمفاوية. وفي الأشخاص المصابين بهذا المرض ، تكون خلايا ما قبل B إما بأعداد صغيرة ، أو قد يعانون من مشاكل في الأداء الوظيفي.
4.2 الفيزيولوجيا المرضية
في حالة عدم وجود بروتين طبيعي ، لا تفرق الخلايا الليمفاوية البائية أو لا تنضج بشكل كامل. بدون الخلايا الليمفاوية B الناضجة ، ستكون خلايا البلازما المنتجة للأجسام المضادة غائبة أيضًا. نتيجة لذلك ، فإن الأعضاء الشبكية البطانية والأعضاء اللمفاوية التي تتكاثر فيها هذه الخلايا وتتمايز وتخزن تكون ضعيفة التطور. قد يتقلص حجم الطحال واللوزتين واللحمية والأمعاء والغدد الليمفاوية الطرفية أو يغيب تمامًا في الأفراد المصابين ب agammaglobulinemia المرتبط بالكروموسوم X.
يمكن أن تؤدي الطفرات في كل منطقة من مناطق الجينات إلى هذا المرض. الحدث الجيني الأكثر شيوعًا هو طفرة الخطأ. تؤدي معظم الطفرات إلى اقتطاع البروتين. تؤثر هذه الطفرات على المخلفات الحرجة في البروتين السيتوبلازمي وهي شديدة التغير وموزعة بالتساوي في جميع أنحاء الجزيء. ومع ذلك ، لا يمكن التنبؤ بخطورة المرض باستخدام طفرات معينة. يؤثر ما يقرب من ثلث الطفرات النقطية على مواقع CGG ، والتي تحتوي عادةً على رمز بقايا الأرجينين.
هذا البروتين المهم ضروري لتكاثر وتمايز الخلايا الليمفاوية البائية. يعاني الرجال المصابون بخلل في البروتين من غياب تام أو شبه كامل للخلايا الليمفاوية في خلايا البلازما.
4.3 الأعراض والمظاهر
تبدأ العدوى المتكررة في الطفولة المبكرة وتستمر طوال فترة البلوغ.
يتمثل المظهر الأكثر شيوعًا لمرض بروتون أو agammaglobulinemia في Bruton في زيادة القابلية للإصابة بالبكتيريا القيحية المغلفة ، مثل عدوى المستدمية النزلية ، وأنواع معينة من الزوائف. تحدث التهابات الجلد في مرضى المرض بشكل رئيسي بسبب المكورات العقدية من المجموعة أ والمكورات العنقودية ، وقد تظهر على شكل قوباء أو التهاب النسيج الخلوي أو خراجات أو دمامل.
قد يظهر شكل من الإكزيما يشبه التهاب الجلد التأتبي ، إلى جانب زيادة في حالات تقيح الجلد الغنغريني ، والبهاق ، والثعلبة ، ومتلازمة ستيفنز جونسون (بسبب زيادة تعاطي المخدرات). تشمل الإصابات الأخرى الشائعة في هذا المرض عدوى الفيروس المعوي ، وتعفن الدم ، والتهاب السحايا ، والإسهال الجرثومي. قد يعاني المرضى أيضًا من أمراض المناعة الذاتية ، قلة الصفيحات ، قلة العدلات ، فقر الدم الانحلاليوالتهاب المفاصل الروماتويدي. نادرًا ما تؤدي العدوى المستمرة بالفيروس المعوي إلى التهاب الدماغ أو التهاب الجلد والعضلات - التهاب السحايا والدماغ. بالإضافة إلى التغيرات العصبية ، تشمل المظاهر السريرية لهذه المتلازمة وذمة وطفح جلدي حمامي على الجلد فوق المفاصل الباسطة.
قد يصاب الرجال بالتهاب الأذن الوسطى والالتهاب الرئوي الحاد و / أو المتكرر بشكل غير عادي. أكثر مسببات الأمراض شيوعًا هو الالتهاب الرئوي S ، يليه فيروس الأنفلونزا B ، والمكورات العنقودية ، والمكورات السحائية ، والموراكسيلا النزلية.
في الأطفال الذين تقل أعمارهم عن 12 عامًا ، تحدث العدوى النموذجية بسبب البكتيريا المغلفة. تشمل الإصابات الشائعة في هذه الفئة العمرية الالتهاب الرئوي المتكرر والتهاب الجيوب الأنفية والتهاب الأذن الوسطى الناجم عن الالتهاب الرئوي S وفيروس الأنفلونزا B ، والتي يصعب علاجها في هذا العمر.
في مرحلة البلوغ ، مظاهر جلديةتصبح أكثر شيوعًا ، عادةً بسبب المكورات العنقودية الذهبية والمكورات العقدية من المجموعة أ. يتم استبدال التهاب الأذن الوسطى بـ التهاب الجيوب الأنفية المزمن، ويصبح مرض الرئة مشكلة مستمرة ، سواء في شكل مقيد أو في شكل انسداد.
يمكن أن يعاني كل من الرضع والبالغين من أمراض المناعة الذاتية. عادةً ما تشمل هذه الاضطرابات التهاب المفاصل ، وفقر الدم الانحلالي المناعي الذاتي ، ونقص الصفيحات المناعي الذاتي ، وقلة العدلات المناعية الذاتية ، ومرض التهاب الأمعاء. قد يكون من الصعب للغاية السيطرة على مرض التهاب الأمعاء وغالبًا ما يساهم في فقدان الوزن المزمن وسوء التغذية. الإسهال شائع وينتج عن أنواع الجيارديا أو العطيفة. المرضى عرضة للإصابة بالفيروس المعوي ، بما في ذلك فيروس شلل الأطفال.
الفحص البدني
قد يكون الأطفال الذكور المصابون بنقص الجلوبيولين في الدم في بروتون أصغر جسديًا من الأطفال الذكور غير المصابين بالمرض بسبب توقف النمو والتطور من العدوى المتكررة.
في الامتحان الغدد الليمفاويةواللوزتين والأنسجة اللمفاوية الأخرى قد تكون صغيرة جدًا أو غائبة تمامًا.
يتم تشخيص المرض عندما يصاب الطفل بالمرض بشكل متكرر في وجود عدوى مختلفة ، التهاب الأذن الوسطى أو عدوى الجلد بالمكورات العنقودية والتهاب الملتحمة التي لا تستجيب للعلاج بالمضادات الحيوية. قد تترافق هذه الالتهابات الشديدة مع قلة العدلات.
تقيح الجلد الغنغريني ، مثل القرح والتهاب النسيج الخلوي الأطراف السفليةيمكن أيضًا أن يؤخذ في الاعتبار عند بعض المرضى.
4.4 التشخيص
يعد الاكتشاف والتشخيص المبكرين ضروريين للوقاية من المراضة والوفاة المبكرة بسبب الالتهابات الجهازية والرئوية. يتم تأكيد التشخيص من خلال مستويات منخفضة بشكل غير طبيعي أو عدم وجود الخلايا الليمفاوية B الناضجة ، وانخفاض أو عدم وجود سلسلة ثقيلة التعبير على سطح الخلايا الليمفاوية. من ناحية أخرى ، سيرتفع مستوى الخلايا اللمفاوية التائية. المحدد النهائي للمرض هو التحليل الجزيئي. يستخدم التحليل الجزيئي أيضًا للتشخيص السابق للولادة ، والذي يمكن إجراؤه بأخذ عينات من الزغابات المشيمية أو بزل السلى عندما يُعرف عن الأم بأنها حاملة للجين المعيب. مستويات IgG أقل من 100 مجم / ديسيلتر تدعم التشخيص.
نادرًا ما يمكن إجراء التشخيص عند البالغين في العقد الثاني من حياتهم. يُعتقد أن هذا ناتج عن طفرة في البروتين بدلاً من غيابه الكامل.
اختبارات المعمل
تتمثل الخطوة الأولى في تحديد كمية IgG و IgM والغلوبولين المناعي E (IgE) والغلوبولين المناعي A (IgA). يجب قياس مستويات IgG أولاً ، ويفضل بعد 6 أشهر من العمر عندما تبدأ مستويات IgG لدى الأمهات في الانخفاض. ثانياً ، مستويات IgG أقل من 100 مجم / ديسيلتر عادة ما تدل على مرض بروتون. كقاعدة عامة ، لا يتم اكتشاف IgM و IgA.
بمجرد تحديد مستوى الجسم المضاد ليكون منخفضًا بشكل غير طبيعي ، سيتم تأكيد التشخيص عن طريق تحليل علامات الخلايا اللمفاوية البائية والخلايا اللمفاوية التائية. مستويات خلايا CD19 + B أقل من 100 مجم / ديسيلتر. تميل قيم فحص الخلايا التائية (CD4 + و CD8 +) إلى الزيادة.
يمكن إجراء مزيد من التحليل عن طريق الكشف عن استجابات IgG لمولدات المضادات المعتمدة على T والمستضدات T المستقلة عن طريق التحصين ، على سبيل المثال ، بعد إعطاء لقاح المكورات الرئوية غير المقترن 23 التكافؤ أو لقاحات الخناق والكزاز والأنفلونزا من النوع B.
يمكن للاختبار الجيني الجزيئي أن يؤسس تأكيدًا مبكرًا لتشخيص تضخم الغاما غلوبولين الدم الخلقي.
اختبارات أخرى
تعد دراسات وظائف الرئة مركزية لرصد أمراض الرئة. يجب إجراؤها سنويًا للأطفال الذين يمكنهم إجراء الاختبار (عادةً من سن 5 سنوات).
إجراءات
يمكن استخدام التنظير الداخلي وتنظير القولون لتقييم المدى والتقدم مرض التهابأمعاء. قد يكون تنظير القصبات مفيدًا في التشخيص والمتابعة مرض مزمنالرئتين والالتهابات.
4.5 العلاج
إن إدخال الغلوبولين المناعي هو الطريقة الرئيسية للسيطرة على المرض. يجب إعطاء جرعات نموذجية من 400-600 مجم / كجم / شهر كل 3-4 أسابيع. يمكن تعديل الجرعات والفترات بناءً على الاستجابات السريرية الفردية. يجب أن يبدأ العلاج في عمر 10-12 أسبوعًا. يجب أن يبدأ العلاج بـ IgG بمستوى لا يقل عن 500-800 مجم / ديسيلتر. يجب أن يبدأ العلاج في عمر 10-12 أسبوعًا.
العلاج المضاد للبكتيريا تتطلب نوبات المضاعفات البكتيرية المعدية في HHHG علاجًا بالمضادات الحيوية ، عادةً بالحقن. الشرط الأساسي لنجاح العلاج المضاد للميكروبات لـ HHHH هو استخدامه في وقت واحد مع نظرية الاستبدالومع ذلك ، في هذه الحالة ، تكون مدة العلاج بالمضادات الحيوية أطول بمقدار 2-3 مرات من مدة العلاج القياسي بالمضادات الحيوية للأعضاء الالتهابية المقابلة المصابة في المرضى المؤهلين مناعياً. تظل جرعات المضادات الحيوية مرتبطة بالعمر ، ولكنها تركز على الالتهابات الشديدة والمتوسطة.
يمكن استخدام سيفترياكسون لعلاج الالتهابات المزمنة أو الالتهاب الرئوي أو تعفن الدم. إذا أمكن ، يجب على الأطباء الحصول على مزارع الحساسية للمضادات الحيوية ، حيث أن العديد من الكائنات الحية تظهر بالفعل مقاومة للعديد من المضادات الحيوية. قد تتطلب عدوى المكورات العقدية ، على وجه الخصوص ، سيفترياكسون أو سيفوتاكسيم أو فانكومايسين.
قد تكون موسعات الشعب الهوائية وأجهزة الاستنشاق بالستيرويد واختبارات وظائف الرئة المنتظمة (3-4 مرات في السنة على الأقل) جزءًا ضروريًا من العلاج بالإضافة إلى المضادات الحيوية.
المظاهر الجلدية المزمنة مرض في الجلديتم التحكم في الأكزيما عن طريق الترطيب اليومي للبشرة باستخدام المستحضرات الخاصة والمنشطات.
جراحة
قد تقتصر الجراحة على الالتهابات الحادة الوخيمة. تشمل الإجراءات الأكثر شيوعًا تلك المستخدمة لعلاج مرضى التهاب الأذن الوسطى المتكرر والمصابين بالتهاب الجيوب الأنفية المزمن.
4.6 المضاعفات
تشمل المضاعفات الالتهابات المزمنة والتهابات الفيروس المعوي المركزي الجهاز العصبي، زيادة في حدوث أمراض المناعة الذاتية ، وكذلك التهابات الجلد. المرضى معرضون بشكل متزايد لخطر الإصابة بسرطان الغدد الليمفاوية.
4.7 الوقاية
نظرًا لأن غاماغلوبولين الدم في بروتون وراثي بطبيعته ، فإن الوقاية منه غير ممكنة. يُنصح بإجراء استشارة وراثية عند التخطيط للحمل للأزواج الذين لديهم تاريخ عائلي لهذا المرض.
استنتاج
مرض بروتون له تشخيص إيجابي مع استمرار إعطاء الغلوبولين المناعي والعلاج المناسب بالمضادات الحيوية. يمكن أن يؤدي الاستخدام غير المناسب للعوامل المضادة للبكتيريا أثناء تفاقم الأمراض المعدية إلى التقدم السريع في العملية المرضية والوفاة.
فهرس
1. أمراض الحساسية والمناعة: المبادئ التوجيهية الوطنية / محرر. ر. خايتوفا ، إن. إيلينا. - م: GEOTAR-media، 2009. - 656 ص.
2. طب الأطفال ، Shabalov N. P. ، St. Petersburg: Spetslit ، 2005.
3. علم المناعة مرحلة الطفولة: دليل عملي لأمراض الطفولة ، أد. A. Yu. Shcherbina و E. D. Pashanova. م: Medpraktika-M، 2006.
استضافت على Allbest.ru
وثائق مماثلة
الأمراض التي يسببها نقص جهاز المناعة. الأمراض الناجمة عن المبالغة في رد فعل الجهاز المناعي. التهابات وأورام الجهاز المناعي. تصنيف نقص المناعة الأولية حسب آليات التطور. تطور مرض بروتون.
عرض تقديمي ، تمت الإضافة بتاريخ 04/19/2013
نقص المناعة الثانوية ، علامات طبيه، الأسباب. فيروس نقص المناعة البشرية ، طرق الانتقال ، المراحل ، العلاج. ملامح مسار الإصابة بفيروس نقص المناعة البشرية عند الأطفال. متلازمة الالتهاب الذاتي ، التصنيف. ملامح تشخيص عدوى فيروس نقص المناعة البشرية الخلقية.
العرض التقديمي ، تمت إضافة 2015/03/15
خصائص جهاز المناعة في الجسم. المناعة المكتسبة وأشكالها. إنتاج الأجسام المضادة وتنظيم إنتاجها. تكوين خلايا الذاكرة المناعية. سمات المناعة المرتبطة بالعمر ، نقص المناعة الثانوية (المكتسبة).
الملخص ، تمت الإضافة بتاريخ 04/11/2010
نقص المناعة كاضطرابات في التفاعل المناعي ؛ تصنيف حالات نقص المناعة. أسباب وميزات IDS الثانوية: النماذج ، عوامل معديةوالأمراض والعلاج. تأثير بيئةعلى حدوث IDS الثانوية.
عرض تقديمي ، تمت إضافة 09/08/2014
الحساسية الذاتية: مفهوم وآليات التنمية. نقص المناعة الأولية والثانوية. عدوى فيروس نقص المناعة البشرية: الجوهر ، المسببات ، المرضية ، آلية المظاهر. المبادئ الممرضة لتصحيح انتهاكات الوظائف الحيوية لجسم الإنسان في الإيدز.
عرض تقديمي ، تمت إضافة 11/11/2014
الجوهر والمسببات والعلامات وتصنيف نقص المناعة الأولية. توطين العيوب الوراثية في PI. المظاهر المعدية المميزة. نقص المناعة الثانوية. مؤشرات لاستخدام بعض الأدوية. الوقاية والعلاج من الالتهابات.
العرض التقديمي ، تمت إضافة 12/21/2014
عوامل الخطر لمرض وراثي. متلازمة "صرخة القطة" وأسبابها وأعراضها. متلازمة ليجون هي مجموعة من التشوهات الخلقية الناتجة عن انتهاك بنية أحد كروموسومات المجموعة ب. الوقاية من الأمراض الوراثية.
عرض تقديمي ، تمت إضافة 04/09/2017
نقص المناعة الأولية: مجتمعة ، T-cell ، B-cell ، عيوب في نظام البالعات وحيدة النواة والخلايا المحببة ، قصور في النظام التكميلي. نقص المناعة الثانوية: فيروسي ، مع أمراض ، واضطرابات استقلابية.
الملخص ، تمت إضافة 08/18/2014
الصورة السريرية لفقر الدم بسبب نقص الحديد. متلازمة فقر الدم الموضوعي. نقص الحديد سابقًا. متلازمة فقر الدم في فقر الدم الضخم الأرومات. كثرة الكريات الصغرية الوراثية (مرض مينكوفسكي شافارد). انتهاك تخليق سبكترين والأنكيرين.
دورة محاضرات تمت الإضافة بتاريخ 07/03/2013
متلازمة نقص المناعة المكتسب (الإيدز). جهاز دفاع الجسم ضد الالتهابات. المصادر الرئيسية للعدوى وطرق انتقال العدوى. خطر الإيدز على جسم الإنسان. استخدام العلاج المضاد للفيروسات القهقرية. الخرافات الأكثر شيوعًا حول الإيدز.