Лечится ли болезнь дюшенна данный время. Дюшенна (–Гризингера) (прогрессирующая) мышечная дистрофия. Физиотерапия и ортопедическая помощь
Относят к группе тяжелых патологий, лечение которых на сегодняшний день представляет трудности. Среди подобных хромосомных аномалий встречаются различные нарушения. Многие из них имеют неврологическую симптоматику. Примерами являются миодистрофия Дюшенна, Беккера. Эти заболевания развиваются еще в детском возрасте и имеют прогрессирующее течение. Несмотря на достижения неврологии, подобные патологии плохо поддаются лечению. Это связано с хромосомными изменениями, которые закладываются в процессе формирования организма.
Описание миодистрофии Дюшенна
Миодистрофия Дюшенна - это генетическое заболевание, проявляющее прогрессирующими нарушениями мышечного аппарата. Патология встречается редко. Распространенность аномалии составляет примерно 3 человека на 10 тысяч лиц мужского пола. Заболевание практически во всех случаях поражает мальчиков. Тем не менее развитие миодистрофии среди девочек не исключено. Данная патология проявляет себя еще в раннем детстве.
Другим заболеванием, имеющим те же причины и симптомы, является миодистрофия Беккера. Она отличается более благоприятным течением. Поражение мышечной ткани наступает значительно позже - в подростковом возрасте. При этом симптомы развиваются постепенно, и больной сохраняет трудоспособность в течение нескольких лет. Как и миодистрофия Дюшенна, данная патология распространена среди мужского населения. Частота встречаемости составляет 1 человек на 20 тысяч мальчиков.

Миодистрофия Дюшенна: нейроиммунология заболевания
Причина обеих патологий кроется в нарушении Х-хромосомы. Генетические изменения, происходящие при миодистрофии Беккера и Дюшенна, изучены еще в 30-х годах прошлого века. Тем не менее этиологическая терапия до сих пор не найдена. Тип наследования аномалии рецессивный. Это означает, что, если патологический ген присутствует у одного из родителей, вероятность рождения больного ребенка составляет 25 %. Х-хромосома является самой длинной в организме. При обоих нарушение происходит в одном и том же локусе (р21). Данное повреждение приводит к снижению синтеза белка, который входит в состав клеточных мембран мышечной ткани. При миодистрофии Дюшенна он полностью отсутствует. Поэтому нарушения проявляются гораздо раньше. При миодистрофии Беккера белок синтезируется в малых количествах или является патологическим.

Клиническая картина миодистрофии
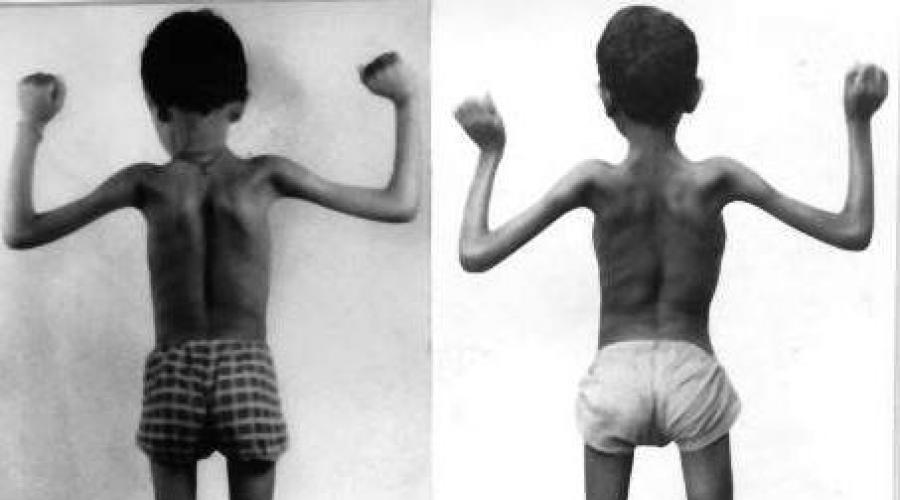
Миодистрофия Дюшенна характеризуется поражением нервно-мышечного аппарата. Заболевание можно заподозрить в возрасте 2-3 лет. В этот период становится заметно, что ребенок отстает в физическом развитии от своих сверстников, плохо ходит, бегает и прыгает. Таким малышам тяжело подниматься по ступеням, она часто падают. Поражение мышц начинается с нижних конечностей. Позже оно распространяется на все проксимальные отделы мускулатуры. Дегенерация происходит в верхнем плечевом поясе, В этих местах наблюдается истончение мускулатуры. С годами миодистрофия прогрессирует. Поражение мышц и постоянная нагрузка на них приводят к контрактурам - стойким искривлениям конечностей. Помимо этого у больных миодистрофией Дюшенна наблюдаются заболевания сердца, которые периодически дают о себе знать. Также для данной патологии характерно снижение интеллектуальных способностей (не сильно выражено).
Миодистрофия Беккера имеет те же симптомы, но развивается позже. Первые проявления наблюдаются в 10-15 лет. Происходит постепенное изменение походки, появляется шаткость, позже развиваются контрактуры. Нарушения со стороны сердечно-сосудистой системы выражены слабо. Интеллект при этом заболевании обычно не снижается.

Как диагностировать миодистрофию?
Диагноз "миодистрофия Дюшенна" (или Беккера) может поставить опытный врач-невролог. В первую очередь он основывается на клинической картине этих заболеваний. Обращают на себя внимание такие симптомы, как истончение мышц проксимальных отделов, ложная гипертрофия икроножной мускулатуры (возникает вследствие фиброза и отложения жировой ткани). Данные проявления практически всегда сочетаются с сердечно-сосудистыми патологиями. На ЭКГ можно заметить нарушение ритма,
Также больные с миодистрофией Дюшенна немного отстают от своих сверстников в умственном развитии. Чтобы это определить, с детьми работает врач-психолог. При подозрении на данное заболевание проводится миография (определение электрического потенциала мускулатуры) и ЭхоКС - исследование камер сердца. Чтобы с точностью определить наличие патологии, выполняется генетическая диагностика. При миодистрофии Беккера и Дюшенна пациенты должны наблюдаться у нескольких специалистов. Среди них - невролог, психолог и кардиолог.

Лечение
К сожалению, этиологическое лечение миодистрофии Дюшенна и Беккера не разработано. Тем не менее больным показана симптоматическая и поддерживающая терапия. На ранних этапах заболевания проводятся курсы лечебной физкультуры, массаж. При значительной инвалидизации необходимо выполнять пассивные движения конечностей. Чтобы замедлить прогрессирование развития разгибательных контрактур, прибегают к фиксации ног во время сна. Поддерживающая терапия позволяет продлить жизнь пациентов и ослабить симптомы заболевания. Используют препараты кальция, медикаменты «Галантамин» и «Прозерин». В некоторых случаях назначают гормональные средства, в основном «Преднизолон». При прогрессирующих нарушениях со стороны сердца назначают кардиопротекторы.
Миодистрофия Дюшенна и Беккера: прогноз
Прогноз миодистрофии Дюшенна неутешителен. Раннее развитие симптомов и быстрое прогрессирование заболевания приводят к инвалидности еще в детском возрасте. Пациентам с данной патологией требуется постоянный уход. В среднем продолжительность жизни больных составляет около 20 лет. Миодистрофия Беккера характеризуется благоприятным течением. При постоянном наблюдении врачей и выполнении их указаний работоспособность больных сохраняется до 30-35 лет.
Дюшенна?
Есть много типов мышечной дистрофии, все они вызваны нарушением генов (единиц наследственности, передаваемых от родителей к детям). При мышечной дистрофии Дюшенна (МДД) недостаток белка дистрофина вызывает ухудшение и разрушение мышц, ведущее к прогрессирующему затруднению ходьбы и общей подвижности. МДД является наиболее часто случающейся и одной из наиболее быстро прогрессирующих детских нейромышечных болезней. Этой болезнью болеет приблизительно каждый 3000-ый новорожденный мальчик в мире. МДД болеют только мальчики (за очень редким исключением).
Как мышечная дистрофия Дюшенна наследуется?
При мышечной дистрофии Дюшенна дефектный ген является Х-сцепленным. Это означает, что этот ген расположен на Х-хромосоме. У женщин две Х-хромосомы, а у мужчин одна Х-хромосома, которую они наследуют от своей матери, и одна Y-хромосома, которую они наследуют от своего отца. Приблизительно в двух третьих случаев дефектный ген передается сыну посредством дефектной Х-хромосомы матери. В этих случаях мать является «носителем», у которого, в большинстве случаев, не проявляются никакие симптомы болезни. Это потому, что этот ген является «рецессивным», а что означает, что ее нормальная Х-хромосома будет доминантной и будет нормально производить дистрофин. Только у очень малого количества носителей наблюдается умеренная степень мышечной слабости, которая обычно ограничивается плечами и бедрами, и такие женщины называются «проявляющимися носителями». Генетическое нарушение могло возникнуть в предыдущем поколении, в котором наблюдалась семейная предрасположенность к этому заболеванию. Однако, приблизительно в одной трети случаев МДД генетическое нарушение возникает в самом мальчике, и тогда оно называется «спонтанной мутацией».
Почему так важно консультирование по вопросам наследственности?
Каждый сын женщины-носителя имеет 50% вероятность унаследовать MДД от дефектной Х-хромосомы его матери, и каждая дочь имеет 50% вероятность стать носителем этой болезни таким же способом. Сразу после диагноза МДД необходимо получить консультацию по вопросам наследственности, а также пройти надлежащее тестирование членам семьи, которые возможно являются носителями. Во время консультации вы получите информацию о последовательности наследственности и об опасности для других членов семьи, а также «прогноз» (возможные последствия болезни). Во время этой консультации также предоставляется информация о диагностическом тестировании, включая предродовое тестирование и тестирование носителя.
Как диагностируется МДД?
Симптомы
Болезнь МДД часто трудно диагностировать, так как симптомы могут быть разными, и если в семье не было этой болезни, наличие МДД может не подозреваться. Довольно обычным считается запаздывание начала ходьбы, когда ребенок делает первые шаги приблизительно в восемнадцать месяцев. При ходьбе больной МДД мальчик может часто падать. Ему часто трудно взбираться по ступенькам, трудно бегать и прыгать, и у него может развиться «утиная» походка. Классическими симптомами является увеличение (гипертрофия) икроножных мышц, которое случается приблизительно в 90% случаев. У него может развиться тенденция ходить на пальцах, что часто сопровождается выпиранием живота и раздвинутыми в коленных суставах ногами, и называется «лордозом». Ему может быть трудно вставать с пола без помощи. Чтобы помочь себе, он может взбираться руками по ногам – это называется «признаком Говерза». Эти симптомы обычно начинают развиваться в возрасте от одного до трех лет и продолжают прогрессировать до тех пор, пока ему не понадобиться кресло-коляска, чаще всего в возрасте от восьми до двенадцати.
Анализ креатинфосфокиназы
Лабораторное тестирование МДД начинается с анализа мышечного энзима, называемого креатинфосфокиназой. Из-за недостатка дистрофина в волокнах мышцы креатинфосфокиназа просачивается из поврежденной мышцы и появляется в крови в больших количествах. Анализ крови может показать уровень креатинфосфокиназы, превышающий нормальный уровень в 50 – 100 раз. Хотя содержание этого энзима часто слегка повышенное при дистрофии других типов (включая ассоциативную мышечную дистрофию Беккера), при МДД его содержание намного выше. Приблизительно у 70% носителей МДД уровень креатинфосфокиназы также будет слегка повышенный. Поэтому высокий уровень креатинфосфокиназы свидетельствует о том, что сами мышцы являются вероятной причиной слабости, но не говорит нам со 100% гарантией, какая мышечная болезнь это может быть.
Изучение ДНК
В настоящее время, для того чтобы установить точный , изучение ДНК ведется с применением новых технологий. Гены состоят из сегментов ДНК (дезоксирибонуклеиновой кислоты), и соответствующие части этого генетического материала могут быть изучены при помощи микроскопа. Аномалии, вызывающие МДД, могут быть трех типов: удаление (отсутствующие части), дублирование (дополнительные части) или точечная мутация (измененные части). Изучение ДНК часто занимает много времени и трудно с технической стороны и, в зависимости от генетического дефекта, могут давать неопределенные результаты. В некоторых случаях эти изучения могут дать точную информацию о генетической аномалии, вызывающей МДД, а в других случаях аномалию невозможно точно определить. Это также относится и к диагностике женщин-носителей. Изучение ДНК может также проводиться до родов в неродившемся ребенке, если в семье было это заболевание.
Биопсия мышц
Если изучение ДНК не дает ясной картины, может потребоваться биопсия мышц. Маленькая частица мышечной ткани, обычно из бедра, извлекается при помощи иглы. Используя специальный метод окрашивания в лаборатории, мышечная ткань изучается при помощи микроскопа на наличие дистрофина. При МДД анализ показывает отсутствие дистрофина, в то время как при родственной болезни мышечной дистрофии Беккера небольшое количество дистрофина присутствует. Поэтому анализ биопсии мышц необходим для установления точного анализа в тех случаях, когда неизвестно, болел ли кто в семье этой болезнью, или когда анализ ДНК не дал определенных результатов.
Только две болезни могут вызвать трудности при диагностировании МДД: мышечная дистрофия Беккера и конечностно-поясная мышечная дистрофия. Вышеупомянутые анализы, в особенности биопсия мышц, могут различать эти болезни.
Вылечивается ли МДД?
В настоящее время МДД не вылечивается, но по всему миру продолжаются широкомасштабные исследования в этой области. Исследователи значительно продвинулись в понимании МДД и продолжают поиски лечения. Некоторыми областями, на которых исследования сконцентрированы в настоящий момент, являются:
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна
(МДД
) - это серьезное рецессивное заболевание, которое характеризуется быстрым прогрессированием мышечной дистрофии
, которая в конечном итоге приводит к полной потере способности двигаться и смерти больного.
Это заболевание поражает примерно 1 человека из 4000, то есть - это наиболее распространенный тип мышечной дистрофии. Обычно на МДД болеют только мужчины, хотя женщины могут иногда быть носителями заболевания. Если же отец болен МДД, а мать является носителем, или тоже больна, то в таком случае на мышечную дистрофию Дюшенна может заболеть женщина. Расстройство возникает в связи с в дистрофин , который у людей расположен на (Xp21). Ген дистрофин кодирует деятельность белка дистрофина, который является важной структурной составляющей мышечной ткани. Дистрофин обеспечивает структурную устойчивость дистрофин-ассоциированного-гликопротеинового комплекса (ДАГ комплекса), расположенного на клеточной мембране.
Симптомы заболевания обычно появляются у детей мужского пола до 5 лет и могут проявиться в раннем детстве. Первыми признаками болезни являются прогрессирующая проксимальная слабость мышц ног и таза, связанная с потерей мышечной массы. Постепенно эта слабость распространяется на руки, шею и другие части тела. Ранние признаки расстройства могут также включать псевдогипертрофию (увеличение икроножных мышц и дельтовидных мышц), низкую выносливость и трудности при стоянии без посторонней помощи, как правило, человек также не может самостоятельно подняться. В процессе прогрессирования заболевания мышечная ткань постепенно заменяется жировой и фиброзной тканями (как следствие развивается фиброз). Для помощи при ходьбе, в возрасте 10 лет может быть необходимым применение специальных подтяжек, но большинство пациентов уже старше 12 лет не могут передвигаться без инвалидной коляски.
Позже возникают следующие признаки расстройства: аномалии развития костей, которые приводят к деформации скелета, в том числе к искривлению позвоночника.
В связи с прогрессивным ухудшением работы мышц, особь теряет возможность двигаться, что в конечном счете, может быть причиной паралича. Относительно отклонений в психическом развитии, то их наличие зависит от каждого конкретного случая, но если определенные отклонения и присутствуют, то они несущественно влияют на развитие ребенка, ведь со временем нарушение не прогрессирует. Средняя продолжительность жизни больных МДД варьирует от подросткового возраста до 20 - 30 лет. Известны случаи, когда больные доживали до 40 лет, но, к сожалению, такие случаи являются скорее исключением.
Распространенность
Мышечная дистрофия Дюшенна возникает в связи с мутациями в гене дистрофин, который расположен на Х-хромосоме. В связи с этим, ДМД встречается у 1 человека на 4000 новорожденных мужского пола. Мутации в гене дистрофин могут быть или возникают спонтанно во время зародышевой линии передачи.
Эпоним
Заболевание названо в честь французского невропатолога Жульема Бенджамина Аманда Дюшенна (Guillaume Benjamin Amand Duchenne), который впервые описал это заболевание в 1861 г оду.
Патогенез
Мышечная дистрофия Дюшенна обусловлена мутацией в гене дистрофин , которого Xp21. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофина приводит к проникновению избыточного кальция в сарколему (клеточную мембрану). Как следствие изменения этих сигнальных путей, вода наполняет митохондрии, которые после этого разрываются. При дистрофии скелетных мышц, митохондриальная дисфункция приводит к усилению стресса вызванного цитозольным-кальциевым сигналом и усилению производства стресс-индуцированных активных форм кислорода (АФК). В этом сложном каскадном комплексе, который включает в себя несколько реакций еще до сих пор не понятно до конца, почему из-за повреждения сарколеммы увеличиваются проявления окислительного стресса, который в итоге приводит к смерти клетки. Мышечные волокна подвергаются некрозу и, наконец, происходит замена мышечной ткани жировой, а также соединительной.
Симптомы
Основным симптомом мышечной дистрофии Дюшенн
а - является мышечная слабость
, которая в первую очередь связана с атрофией мышц, а именно скелетной мышечной ткани. В первую очередь атрофируются мышцы бедер, таза, плеч и икроножные мышцы. Мышечная слабость возникает также в руках, шее и других частях тела, но обычно не так рано, как в нижней части тела. Икры часто увеличены. Обычно симптомы появляются в возрасте до 6 лет, но могут впервые проявиться еще в раннем детстве.
Другие физические симптомы
расстройства:
- неуклюжая (тяжелая) походка, шаги или бег (как правило, пациенты, ходят на пальцах (на носочках), через повышенный тонус икроножных мышц). Кроме того, такая манера ходить, является своеобразной адаптацией к постепенной потере функций колен;
- больные часто падают;
- постоянная усталость;
- у больного возникают трудности при осуществлении таких двигательных навыков как бег или скачки;
- усиление поясничного лордоза, которое приводит к атрофии (уменьшение размеров) мышц сгибателей бедра. Она влияет, в общем, как на осанку, так и на манеру ходить и бегать, в частности;
- мышечные контрактуры, которые существенно уменьшают функциональность ахилового и подколенного сухожилия, поскольку количество мышечных волокон уменьшаются и возникает фиброз мышц;
- прогрессирование трудности при ходьбе;
- деформация мышечных волокон;
- дсевдогипертрофия (увеличение) языка и икроножных мышц, вызванная заменой мышечной ткани жировой и соединительной;
- повышенный риск нейроповеденческого расстройства (такого как синдром дефицита внимания и гиперактивности (СДВГ), расстройства аутистического спектра), трудности с учебой (дислексия) и не прогрессирующие отклонения в определенных когнитивных функциях (в частности, таких как краткосрочная словесная память), которые, как считают ученые, возникают из-за отсутствия или нарушения функционирования дистрофина в мозге;
- возможная потеря способности ходить (обычно в возрасте до 12 лет);
- скелетные деформации (в некоторых случаях возникает сколиоз);
Признаки и тестирования
Как уже было сказано, атрофия мышц при МДД начинается в виде мышечной слабости в ногах и тазовом поясе, затем переходит к мышцам плеч и шеи, после чего, повреждает мышцы рук и дыхательные мышцы. Важным видимым признаком в начале развития заболевания является увеличение икроножных мышц (псевдогипертрофии ). Распространенным явлением есть кардиомиопатия, но развитие сердечной недостаточности или аритмии (заболевания связаны с нарушениями ритма сердца, последовательности и силы сокращений сердечной мышцы) встречаются довольно редко.
Наличие симптома Говерса отражает более тяжелые нарушения мышц нижних конечностей. Можно говорить о наличии симптомов, в случае, если ребенок помогает себе встать с помощью рук: сначала, ребенок становится на четвереньки (опираясь на пол ногами и руками), а затем, держась руками за ноги, контролирует направление своего движения;
- больные МДД дети, часто устают быстрее и имеют меньше силы, чем их сверстники;
- очень высокий уровень креатин-киназы (КФК-ММ) в крови тоже может стать показателем развития и прогрессирования заболевания;
- при проведении электромиографии (ЭМГ) видно, что слабость организма вызвана повреждением мышечной ткани, а не повреждением нервной проводимости;
- может выявить генетические нарушения в гене Xp21;
- мышечная биопсия с последующим гистологическим, иммуногистохимическим или имуноблотинговим исследованием) или генетическое тестирование (с помощью анализа крови), подтверждает отсутствие дистрофин.
Диагностика
ДНК-тест
Мышечно-специфическая изоформа гена дистрофина состоит из 79 экзонов. Тестирование и их анализ, как правило, позволяют определить тип мутации экзона или определить какие экзоны повреждены. Анализ ДНК в большинстве случаев подтверждает предварительную диагностику другими методами.
Биопсия мышц
Если при анализе ДНК никаких мутаций не обнаруживается, то возможно проведение мышечной биопсии. Для этой процедуры с помощью специального инструмента берут маленький образец мышечной ткани и, использовав специальный краситель, определяют наличие /отсутствие в мышечной ткани дистрофина. Полное отсутствие белка указывает на наличие этого заболевания.
За последние несколько лет ДНК-тесты были существенно усовершенствованы, на сегодня они проявляют больше мутаций и поэтому мышечную биопсию для подтверждения МДД сейчас используют все реже.
Пренатальное тестирование
Если один или оба родителя являются "носителями" этого заболевания, то существует риск того, что их еще не народившийся ребенок будет поражен этим расстройством. Для определения того, будет будущий ребенок больным МДД, используют методы . На сегодня эти методы доступны только для определения некоторых нервно-мышечных расстройств. Различные пренатальные тесты могут проводиться примерно на 11 недели беременности.
Исследование с помощью биопсии хориона
(CVS) можно проводить на 11-14 неделях, амниоцентез
можно использовать после 15 недели, забор крови плода
возможен примерно на 18 неделе. Родители должны внимательно изучить все возможные методы и, возможно, с помощью выбрать наиболее оптимальный для себя вариант. Если тестирование будет осуществлено на ранних сроках беременности, то это позволит досрочно прекратить беременность, в случае наличия заболевания у плода, однако, при применении таких методов, увеличивается риск выкидыша при последующих беременностях, чем при тех методах, которые применяются позже (около 2% , по сравнению с 0,5%).
Лечение
Никаких известных эффективных препаратов для лечения мышечной дистрофии Дюшенна, не существует. Хотя согласно последним исследованиям стволовых клеток существуют перспективные векторы, которые могут заменить поврежденные мышечные ткани. Однако, на данном этапе лечения, как правило, симптоматическое и направлено на улучшение качества жизни больного человека.
Оно включает в себя:
Употребление таких кортикостероидов как преднизолон и дефлазакорт для увеличения энергии и сил и облегчения тяжести некоторых симптомов;
- рандомизированные контролируемые исследования показывают, что использование бета 2-агонистов увеличивает мышечную силу, но не замедляет процесс прогрессирования заболевания. Время контроля за людьми, которые употребляли бета 2-агонисты составляет около 12 месяцев, следовательно, результаты этих испытаний не могут быть экстраполированы на больший период времени;
- рекомендуется умеренная физическая активность, разрешается заниматься плаванием. Бездействие (например, постельный режим) может усилить прогрессирование заболевания;
- для поддержания мышечной силы, гибкости и функциональности суставов важна физиотерапия;
- использование ортопедических приспособлений (например, инвалидных колясок) может улучшить способность больного двигаться и самостоятельно обеспечивать свои потребности. Использование так называемых съемных стяжек, фиксирующих голень во время сна позволяет отложить начало контрактур (ограничение движений суставов).
- по мере прогрессирования заболевания необходимым становится использование специальных респираторных механизмов, позволяющих обеспечить нормальный процесс дыхания.
Центром по контролю и профилактике заболеваний (Centers for Disease Control and Prevention (CDC)) были разработаны общие многопрофильные стандарты (принципы) помощи больным МДД. Эти принципы были опубликованы в двух частях в журнале The Lancet Neurology в 2010 году. (Http://www.treat-nmd.eu/patients/DMD/dmd-care).
Прогноз
Мышечная дистрофия Дюшенна повреждает все скелетные мышцы, мышцы сердца и дыхательные мышцы (на более поздних стадиях). Больные МДД, как правило, живут только к подростковому возрасту или умирают в возрасте 30-40 лет. Последние достижения в области медицины, позволяют надеяться на увеличение продолжительности жизни больных этим расстройством.
Иногда (но очень редко) особи с МДД доживали до 40-50 лет, но лишь с помощью использования надлежащего дополнительного оборудования (инвалидных колясок и кроваток), вентиляционной поддержки дыхания (с помощью трахеостомии или специальной дыхательной трубки), очистки дыхательных путей и принятие необходимых сердечных препаратов. Кроме того, для увеличения продолжительности жизни необходимо на ранних этапах заболевания спланировать механизм ухода за больным на более поздних этапах.
Физиотерапия
Физиотерапия при МДД в основном направлена на больных детей и на развитие их максимального физического потенциала. Цель физиотерапии состоит в следующем:
Минимизировать развитие контрактур и деформаций путем разработки соответствующей программы для сохранения эластичности мышц, а также возможна разработка программы физических упражнений;
Предусмотреть и минимизировать появление вторичных физических осложнений;
Контролировать дыхательные функции и предоставление информации относительно того, какие техники и методы дыхательных упражнений необходимо применять, чтобы очистить дыхательные пути от выделений;
Механическая вентиляция (респираторная помощь)
Использование современных аппаратов искусственной вентиляции легких, которые доставляют регулируемый объем (количество) воздуха в легкие человека, особенно важно это для людей, страдающих от дыхательных проблем, возникающих в процессе развития мышечной дистрофии. Применение этих механизмов при заболевании МДД можно начать в подростковом возрасте, когда дыхательные мышцы начинают повреждаться. Однако, известны случаи, когда даже в возрасте 20 лет, больные не нуждались в использовании таких аппаратов.
Если жизненная емкость легких упала ниже 40% от нормы, то эти респираторы могут использоваться во время сна, ведь именно в это время заболевший может больше всего пострадать от гиповентиляции .
Гиповентиляция во время сна определяется тщательным исследованием истории этого расстройства сна, путем проведения оксиметрии и измерением количества углекислого газа в капиллярной крови. Для вентиляции необходимым может быть проведение процедуры интубации или трахеотомии трубки, через которые воздух непосредственно доставляется в легкие, однако, для некоторых людей вполне достаточно того воздуха, который поступает при использовании специальной маски.
Если жизненная емкость легких продолжает снижаться и составляет менее 30% от нормы, то необходимым становится увеличение продолжительности использования аппарата искусственной вентиляции легких (эта продолжительность должна увеличиваться, по мере необходимости). Трахеотомическая трубка может использоваться как в дневное время, так и во время сна, однако возможна также ситуация, когда достаточным является и количество воздуха, поступающего через дыхательную маску. Аппарат искусственной вентиляции легких легко помещается на вентиляторном лотке снизу или сзади инвалидной коляски, для большей портативности этот механизм можно обеспечить специальной энергетической батареей.
Проводимые исследования
Для выявления лекарств, которые бы позволили смягчить последствия действия МДД или же вообще вылечить ее сегодня ведутся весьма перспективные исследования. Существует много направлений этих исследований, особенно стоит отметить лечения стволовыми клетками, технологию пропуска-экзонов, аналоговую активацию и генную замену. Другим направлением исследований является поддерживающая терапия, которая направлена на разработку лекарств, которая бы предотвратить развитие и прогрессирование болезни.
Лечение стволовыми клетками
Ученые считают, что стволовые клетки, выделенные из мышц (клетки-сателлиты) имеют способность превращаться в миоциты. При непосредственном введении в мышцы животных, они не могут распространяться по всему организму. И для того, чтобы такая терапия была эффективной, необходимо осуществлять инъекции в каждую мышцу через каждые 2 мм. Этот недостаток лечебной процедуры можно исправить, использовав другие, мультипотентные стволовые клетки, которые называются перициты. Они расположены в кровеносных сосудах скелетных мышц.
Эти клетки можно ввести в организм системно и усваиваются они организмом путем попадания в кровоток. Попав, в сосудистую сетку перициты сливаются, образуя миотубулы. Это означает, что они могут быть введены артериально, далее они попадают через стенки сосудов в мышцы. Эти данные свидетельствуют о наличии потенциала для проведения клеточной терапии МДД. Небольшое количество перицитов может быть получена из организма человека, затем они могут быть выращены искусственным путем и введены в кровоток после чего, как считают исследователи, существует вероятность того, что они могут найти «свой путь» к пораженным мышцам.
Активация утрофина
Регулирования экспрессии утрофина для лечения МДД представляет большой интерес, поскольку именно этот является ближайшим эндогенным аналогом дистрофина в . Этот ген короче и расположен у человека на . Исследователи в настоящее время сфокусированы на понимании регулирования за свое выражение в клетках. Еще ранее стало известно, что активация утрофину может частично компенсировать недостаток дистрофин в мышечных клетках. Последние лабораторные исследования утрофину показали существенное улучшение роста мышц у мышей с ДМД. Дальнейшие исследования, которые будут проводиться с участием людей могут дать ответ на вопрос относительно того, действительно активация утрофину у людей больных МДД повысит качество и продолжительность их жизни.
Нуклеотиды были использованы для коррекции нарушений сплайсинга в клетках, полученных от лиц, больных бета-талассемией и использованы для исследования их действия при лечении МДД, спинальной мышечной атрофии, синдрома Хатчинсона-Гилфорда и других заболеваний.
Для лечения лиц больных МДД, использование AONs, согласно исследованиям может быть очень перспективным. Например, МДД может возникать в результате изменений мРНК, вызванных сдвигом рамки считывания (например, вставки или сплайс-мутаций). Предполагается, что в случае если заболевание вызвано, этими нарушениями, то оно может быть излечено путем восстановления последовательности мРНК, то есть возвращение рамки считывания на нужное место. Для того чтобы это сделать необходимо, чтобы AON-и помогли выявить определенные регионы пре-мРНК, которые бы помогли замаскировать распознавания Сплайсосома экзона или экзонов.
И хотя использование AON-ов - может быть достаточно перспективным, но одной из основных проблем является их постоянное возвращение в мышцы. Методы постоянного системного поступления на сегодня испытываются на людях.
Кроме того, исследуются также новые методы, которые бы позволили обойти все недостатки выше описанной процедуры. Эта терапия состоит в изменении U7 малой ядерной РНК в 5 положении еще до в целевых регионах пре-мРНК. Этот метод действует на мышах, пораженных МДД.
Мышечная дистрофия Дюшенна – это генетическое заболевание, связанное с нарушением строения мышечных волокон. Мышечные волокна при этой болезни, в конце концов, распадаются, и теряется способность к передвижению. Мышечная дистрофия Дюшенна передается сцеплено с полом, болеют лица мужского пола. Проявляет себя уже в детском возрасте. Помимо мышечных нарушений, заболевание приводит к скелетным деформациям, может сопровождаться дыхательной и сердечной недостаточностью, умственными и эндокринными нарушениями. Радикального способа лечения, позволяющего искоренить болезнь, пока нет. Все существующие меры являются лишь симптоматическими. Довольно редко больным удается пережить рубеж 30 лет. Эта статья посвящена причинам, симптомам, диагностике и лечению мышечной дистрофии Дюшенна.
Заболевание впервые описано в 1861 году (по другим данным – 1868 году) французским невропатологом и носит его имя. Встречается не так уж и редко: 1 случай на 3500 новорожденных детей. Из всех известных медицине мышечных дистрофий является наиболее распространенной.
Причина заболевания

В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Заболевание наследуется по рецессивному типу, сцепленному с Х хромосомой. Что это означает? Поскольку все гены человека парные, то есть дублируют друг друга, то для того, чтобы появились патологические изменения в организме при наследственном заболевании, необходимо, чтобы генетический дефект возник в одной хромосоме или аналогичных участках обеих хромосом. Если заболевание возникает только при мутациях в обеих хромосомах, то такой тип наследования называют рецессивным. Когда же генетическая аномалия выявляется только в одной хромосоме, но болезнь все равно развивается, такой тип наследования называют доминантным. Рецессивный тип возможен только при одновременном поражении идентичных хромосом. Если вторая хромосома будет «здорова», то заболевание не возникнет. Именно поэтому мышечная дистрофия Дюшенна является уделом лиц мужского пола, потому что они имеют в генетическом наборе одну Х хромосому, а вторую (парную) – У. Если мальчику попадается «поломанная» Х хромосома, то у него обязательно возникает болезнь, потому что здоровой хромосомы у него просто нет. Для того, чтобы мышечная дистрофия Дюшенна возникла у девочки, необходимо совпадение в ее генотипе двух патологических Х хромосом, что практически маловероятно (в таком случае папа девочки должен быть болен, а у мамы в генетическом наборе должна содержаться дефектная Х хромосома). Девочки выступают лишь носителями заболевания и передают его своим сыновьям. Конечно, часть случаев заболевания не является результатом передачи по наследству, а возникает спорадически. Это означает появление мутации в генетическом наборе ребенка спонтанно. Вновь появившаяся мутация может быть передана по наследству (при условии сохранения способности к размножению).
Симптомы заболевания
Мышечная дистрофия Дюшенна всегда заявляет о себе до 5-летнего возраста. Наиболее часто первые симптомы возникают еще до 3-х лет. Все патологические проявления заболевания можно разделить на несколько групп (в зависимости от характера изменений):
- поражение скелетной мускулатуры;
- деформации скелета;
- поражение сердечной мышцы;
- умственные нарушения;
- эндокринные расстройства.
Поражение скелетных мышц

Поражение мышечной ткани является основным проявлением заболевания. Оно становится причиной генерализованной мышечной слабости. Начальные симптомы подкрадываются незаметно.
Рождаются дети без особых отклонений. Однако их двигательное развитие отстает в темпах по сравнению со сверстниками. Такие дети менее активные и подвижные в двигательном плане. Пока ребенок совсем мал, это часто связывают с особенностями темперамента и не обращают внимания на начальные изменения.
Явные признаки возникают с началом ходьбы. Дети часто падают и передвигаются на пальцах (на носочках). Следует отметить, что эти нарушения интерпретируют не при первых шагах ребенка, потому что прямохождение вначале для всех детей сопряжено с падениями и неуклюжестью. Когда большинство ровесников уже вполне уверенно передвигается, мальчики с мышечной дистрофией Дюшенна упорно продолжают падать.
Когда ребенок научится разговаривать, он начинает жаловаться на слабость и быструю утомляемость, непереносимость физических нагрузок. Бег, лазанье, прыжки и другие любимые виды активной деятельности детей для ребенка с мышечной дистрофией Дюшенна не привлекательны.
Походка таких детей напоминает утиную: они как бы переваливаются с ноги на ногу.
Своеобразным проявлением заболевания является симптом Говерса. Он заключается в следующем: при попытке ребенка подняться с колен, корточек, с пола он пользуется руками, чтобы помочь слабым мышцам ног. Для этого он опирается руками на себя, «взбираясь по лесенке, по самому себе».
Мышечная дистрофия Дюшенна имеет восходящий тип мышечной слабости. Это означает, что сначала слабость проявляет себя в ногах, затем распространяется на таз и туловище, потом на плечи, шею и, в конце концов, на руки, дыхательную мускулатуру и голову.
Несмотря на то, что при этом заболевании мышечные волокна подвергаются разрушению и развитию атрофии, внешне некоторые мышцы могут выглядеть вполне нормальными или даже накаченными. Развивается так называемая псевдогипертрофия мышц. Чаще всего этот процесс заметен в икроножных, ягодичных и дельтовидных мышцах, мышцах языка. Место распавшихся мышечных волокон занимает жировая ткань, именно поэтому создается эффект хорошей развитости мускулатуры, что на проверку оказывается совсем не так.
Атрофический процесс в мышцах всегда симметричный. Восходящее направление процесса приводит к возникновению «осиной» талии, «крыловидных» лопаток (лопатки отстают от туловища, словно крылья), симптома «свободных надплечий» (когда голова как бы проваливается в плечи при попытке поднять ребенка под мышки). Лицо гипомимично, губы могут утолщаться (замена мышц жировой и соединительной тканью). Псевдогипертрофия языка становится причиной речевых расстройств.
Разрушение мышц сопровождается развитием мышечных контрактур и укорочением сухожилий (хорошо заметно на примере ахиллового сухожилия).
Сухожильные рефлексы (коленный, ахиллов, с бицепса, с трицепса и так далее) постепенно снижаются. Мышцы на ощупь плотные, но безболезненные. Мышечный тонус обычно снижается.
Постепенное прогрессирование мышечной слабости приводит к тому, что к 10-12 годам многие дети утрачивают способность самостоятельно передвигаться и нуждаются в инвалидном кресле. Способность стоять сохраняется, в среднем, до 16 лет.
Отдельно следует сказать о вовлечении в патологический процесс дыхательной мускулатуры. Это наблюдается после подросткового периода. Слабость диафрагмы и других мышц, участвующих в акте дыхания, приводит к постепенному снижению жизненной емкости легких и объемов вентиляции. По ночам это особенно заметно (появляются приступы удушья), поэтому у детей могут возникать страхи перед сном. Формируется дыхательная недостаточность, которая усугубляет течение интеркуррентных инфекций.
Деформации скелета

Это сопутствующие мышечным изменениям симптомы. У детей постепенно формируются усиление поясничного изгиба (лордоза), искривление грудного отдела позвоночника в сторону (сколиоз) и сутулость (кифоз), меняется форма стопы. Со временем развивается диффузный остеопороз. Эти симптомы еще больше способствуют ухудшению двигательных нарушений.
Поражение сердечной мышцы
Является обязательным симптомом мышечной дистрофии Дюшенна. У больных развивается кардиомиопатия (гипертрофическая или дилатационная). Клинически это проявляет себя нарушениями сердечного ритма, перепадами артериального давления. Границы сердца увеличиваются, однако такое большое сердце имеет малые функциональные возможности. В конце концов, формируется сердечная недостаточность. Сочетание выраженной сердечной недостаточности с дыхательными расстройствами на фоне присоединившейся инфекции может быть причиной смертельного исхода у больных с мышечной дистрофией Дюшенна.
Умственные нарушения
Это не обязательный, но возможный признак заболевания. Он связан с дефицитом особой формы дистрофина – аподистрофином, содержащимся в головном мозге. Нарушения интеллекта варьируются от незначительных до степени идиотии. При этом выраженность умственных нарушений никоим образом не связана со степенью мышечных расстройств. Социальная дезадаптация из-за невозможности свободно передвигаться и посещать детские учреждения (садики, школы) способствует усугублению когнитивных расстройств.
Эндокринные расстройства
Встречаются у 30-50% больных. Могут быть довольно разнообразными, но чаще всего это ожирение с преимущественным отложением жира в области молочных желез, бедер, ягодиц, плечевого пояса, недоразвитие (или нарушение функции) половых органов. Больные часто имеют низкий рост.
Мышечная дистрофия Дюшенна неуклонно прогрессирует. К 15-20 годам почти все больные не в состоянии себя обслуживать самостоятельно ввиду обездвиженности. В конце концов, присоединяются бактериальные инфекции (органов дыхания и мочевыделительной сферы, инфицированные пролежни при недостаточном уходе), которые на фоне сердечной и дыхательной недостаточности приводят к смертельному исходу. Мало кто из больных переживает рубеж в 30 лет.
Диагностика

Диагностика мышечной дистрофии Дюшенна основывается на нескольких видах исследований, основным из которых является генетический тест (ДНК-диагностика).
Только обнаружение дефекта Х хромосомы в том участке, который отвечает за синтез дистрофина, достоверно подтверждает диагноз. До проведения такого анализа диагноз является предварительным.
Из других методов исследования могут применяться:
- определение активности креатинфосфокиназы (КФК). Этот фермент отражает гибель мышечных волокон. Его концентрация при мышечной дистрофии Дюшенна превышает норму в десятки и сотни раз до 5-летнего возраста. Позже уровень фермента постепенно снижается, потому что часть мышечных волокон уже необратимо разрушена;
- электромиография. Этот метод позволяет подтвердить тот факт, что в основе заболевания лежат первичные изменения мышц, а нервные проводники при этом совершенно интактны;
- биопсия мышц. С ее помощью определяют содержание белка дистрофина в мышце. Однако в связи с усовершенствованием генетической диагностики в последние десятилетия эта травматичная процедура отошла на второй план;
- дыхательные пробы (исследование жизненной емкости легких), ЭКГ, УЗИ сердца. Эти методы не используются для установления диагноза, но необходимы для выявления патологических изменений со стороны дыхательной и сердечно-сосудистой систем для того, чтобы скорригировать имеющиеся нарушения.
Выявление больного ребенка в семье означает, что в генотипе матери имеется патологическая Х хромосома. В редких случаях мать может быть здоровой, если мутация возникла у ребенка случайно. Наличие дефектной Х хромосомы несет в себе риск для последующих беременностей. Поэтому такие семьи должен консультировать генетик. При наступлении повторных беременностей родителям предлагают пренатальную диагностику, то есть исследование генотипа еще не родившегося ребенка с целью исключения наследственных заболеваний, в том числе мышечной дистрофии Дюшенна.
Для исследования понадобятся клетки плода, которые получают с помощью различных процедур на разных сроках беременности (например, биопсия хориона, амниоцентез и другие). И хотя эти медицинские манипуляции несут в себе определенный риск для беременности, они позволяют точно ответить на вопрос: имеется ли у плода генетическое заболевание.
Лечение
Мышечная дистрофия Дюшенна является в настоящее время неизлечимым заболеванием. Можно помочь ребенку (взрослому) продлить время двигательной активности, с помощью различных способов поддерживая мышечную силу, компенсируя изменения со стороны сердечно-сосудистой и дыхательной систем.
Несмотря на это, прогнозы ученых в отношении полного излечения от этого заболевания довольно оптимистичны, поскольку уже сделаны первые шаги в этом направлении.
В настоящее время, для лечения мышечной дистрофии Дюшенна из медикаментозных препаратов используют:
- стероиды (при регулярном применении они позволяют уменьшить мышечную слабость);
- β-2-адреномиметики (также временно придают силу мышцам, но не замедляют прогрессирование заболевания).
Применение β-2-адреномиметиков (Альбутерол, Формотерол) не имеет статистически достоверного признания, поскольку существует небольшой опыт их использования при данной патологии. Контроль изменений состояния здоровья у группы пациентов, применявших эти препараты, проводился в течение одного года. Поэтому утверждать, что они работают более длительное время, нет возможности.
Основой лечения сегодня являются стероиды. Считается, что их использование позволяет какое-то время сохранять мышечную силу, то есть они могут затормозить прогрессирование болезни. Кроме того, доказано, что стероиды уменьшают риск возникновения сколиоза при мышечной дистрофии Дюшенна. Но все же возможности этих препаратов ограничены, и заболевание будет неуклонно прогрессировать.
 Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Из медикаментозных препаратов также при мышечной дистрофии Дюшенна применяют сердечные средства (антиаритмические, метаболические, ингибиторы ангиотензинпревращающего фермента). Они позволяют бороться с кардиологическими аспектами заболевания.
Из немедикаментозных методов лечения значимую роль играют физиотерапия и ортопедическая помощь. Физиотерапевтические методики позволяют дольше, чем без их применения, сохранять гибкость и подвижность суставов, поддерживают мышечную силу. Доказано, что умеренная физическая активность благоприятно влияет на течение заболевания, а вот бездействие и постельный режим наоборот способствуют еще более быстрому прогрессированию заболевания. Поэтому необходимо как можно дольше поддерживать посильную физическую деятельность, даже после того, как больной «пересел» в инвалидное кресло. Показаны регулярные курсы массажа. На самочувствие больного положительно влияет плавание.
Ортопедические приспособления позволяют значительно облегчить жизнь больного. Их перечень довольно широк и разнообразен: это и различного рода вертикализаторы (помогают сохранять положение стоя), и приспособления для самостоятельного вставания, и инвалидные кресла-коляски с электрическим приводом, и специальные шины для устранения контрактур в голени (используются даже ночью), и корсеты для позвоночника, и длинные шины для ног (колено-голеностопные ортезы), и многое другое.
Когда заболевание поражает и дыхательные мышцы, и самостоятельное дыхание становится неэффективным, то возможно применение аппаратов искусственной вентиляции легких различной модификации.
И все же даже использование всех этих мер в комплексе не позволяет побороть заболевание. На сегодняшний день, есть ряд перспективных направлений исследований, которые, возможно, станут прорывом в лечении мышечной дистрофии Дюшенна. К наиболее распространенным среди них относят:
- генную терапию (введение «правильного» гена с помощью вирусных частиц, доставку генетических конструкций в составе липосом, олигопептидов, полимерных носителей и другие);
- регенерацию мышечных волокон с помощью стволовых клеток;
- трансплантацию миогенных клеток, которые способны к синтезу нормального дистрофина;
- пропуск экзонов (с помощью антисмысловых олигорибонуклеотидов) в качестве попытки замедлить прогрессирование заболевания и смягчить его течение;
- замену дистрофина на другой белок атрофин, ген которого расшифрован. Методика опробована на мышах и дала положительные результаты.
Каждая из новых разработок несет в себе надежду для больных с мышечной дистрофией Дюшенна на полное выздоровление.
Таким образом, мышечная дистрофия Дюшенна – это генетическая проблема лиц мужского пола. Болезнь характеризуется прогрессирующей мышечной слабостью из-за разрушения мышечных волокон. В настоящее время является неизлечимым заболеванием, однако многие ученые мира трудятся над созданием радикального способа борьбы с ним.
Мультипликационный фильм «Мышечная дистрофия Дюшенна», англ. озвучка, субтитры на русском языке:
Мышечная дистрофия Дюшенна (миопатия) считается крайне тяжёлой наследственной болезнью с прогрессирующим течением, для которой характерно первичное поражение мышц. Это заболевание известно с середины позапрошлого столетия, когда невролог Гийом Дюшенн провёл комплексный анализ мышечной патологии и представил его научному сообществу. Выделяют несколько вариантов течения болезни, которые выделены в отдельные нозологические формы.
Миопатия Дюшенна фиксируется у одного младенца из 4 тысяч новорождённых детей. Среди всех классифицированных мышечных дистрофий эта форма считается наиболее распространённой.
Причины
Заболевания связывают с мутацией гена DMD, отвечающего за выработку белка дистрофина. Этот ген располагается на X-хромосоме. Основная функция белка дистрофина заключается в обеспечении структурной устойчивости специфического гликопротеинового комплекса, который находится на базальной мембране мышечной клетки. Как правило, миопатией Дюшенна страдает мужской пол. В то же время женщины могут быть носителями болезни.
Клиническая картина
Миопатия Дюшенна начинает проявляться у мальчиков до 5 лет. У ребёнка наблюдается быстрая утомляемость. Он часто падает, ему достаточно трудно подняться даже по лестнице. Какие клинические симптомы будут характерны:
- Прогрессирующая слабость в ногах.
- «Утиная» походка. При ходьбе старается опираться на передний отдел стопы.
- Со временем слабость в мышцах переходит на верхние конечности, шею, торс.
- Выявляется псевдогипертрофия. Икроножные и дельтовидные мышцы увеличены в размерах за счёт жировой и соединительной ткани.
- Низкая выносливость.
- Контрактуры (ограничение подвижности) в суставах рук и ног.
- Тяжело стоять без посторонней помощи.
- С большим трудом поднимается с кровати.
- В 8–10 летнем возрасте уже не могут самостоятельно ходить.
- Выраженные искривления позвоночного столба.
- Прогрессирующая мышечная дистрофия приводит к развитию паралича.
- Примерно с 12 лет практически все пациенты не могут обойтись без инвалидной коляски.
Довольно-таки рано отмечается поражение миокарда. Дети жалуются на одышку и появление болезненных ощущений в области сердца. Обычно летальный исход связан с тяжёлыми проблемами с дыхательной системой и сердцем. Средняя продолжительность жизни пациентов варьирует от 20 до 30 лет. Встречаются единичные случаи, когда люди с мышечной дистрофией доживали до 40 лет.
У большинства больных серьёзных психических отклонений не обнаруживается, но всё зависит от индивидуальных особенностей и наследственной предрасположенности.
Диагностика

Характерная клиническая картина предоставляет весомые основания заподозрить мышечную дистрофию. Лабораторно-инструментальная диагностика заболевания состоит из следующих методов:
- ДНК-тест.
- Электромиография.
- Биопсия мышечных волокон.
- Пренатальная диагностика.
Благодаря новейшим технологиям можно провести генетическое тестирование, которое позволяет выявить мутации. В превалирующем большинстве случаев молекулярно-генетический анализ подтверждает результаты других методов диагностики. Электромиография даёт возможность оценить состояние скелетных мышц и сделать вывод, что слабость обусловлена поражением мышечных волокон, а не нарушением нервной проводимости.
Если генетическое тестирование не выявило мутаций, то могут прибегнуть к проведению биопсии мышечных волокон. В процессе этой манипуляции берут совсем небольшой образец ткани и проводят гистологическое исследование. При не обнаружении в мышечной ткани белка дистрофина, можно с достаточно высокой вероятностью утверждать, что у пациента мышечная дистрофия Дюшенна. Следует отметить, что современные ДНК-тесты стали более точными, и биопсию мышечных волокон применяют всё реже.
В случае, когда мать и отец являются носителями мутационного гена, весьма высок риск рождения ребёнка с этой наследственной патологией. Имеется ли наследственный дефект у плода – это можно определить с помощью методов пренатальной диагностики:
- Биопсия хориона проводится на 11–14 неделях.
- Амниоцентез допустим после 15 неделе.
- Взять кровь у плода возможно на 18 неделе.
При выборе того или иного метода пренатальной диагностики следует руководствоваться рекомендациями врача-генетика. Проведение специальных исследований на ранних сроках вынашивания плода позволяет своевременно прервать беременность в случае выявления наследственной патологии. Вместе с тем, используя эти методы диагностики, повышается риск развития выкидыша в дальнейшем.
Ведущим клиническим симптомом миопатии Дюшенна является прогрессирующая мышечная слабость, обусловленная атрофическими изменения в мышцах.
Лечение

К сожалению, на сегодняшний день эффективного лечения, которое поможет избавить пациента от наследственной миопатии Дюшенна, не существует, также как и от . Учитывая результаты последних клинических исследований, большие надежды возлагают на применение стволовых клеток, которые должны будут заменить патологические мышечные волокна. Тем не менее сейчас лечение носит симптоматический характер, и его основная задача постараться улучшить качество жизни пациента. Какие лечебные методы применяются:
- Медикаментозная симптоматическая терапия.
- Поддержка дыхательной функции.
- Использование различных ортопедических средств (фиксирующие ремни, и др.).
- Физиотерапевтические процедуры.
- Массаж.
- Лечебная физкультура.
Несмотря на все старания современной медицины, миопатия Дюшенна остаётся неизлечимым заболеванием.
Симптоматическая терапия
При использовании медикаментозного лечения отмечается положительная динамика в течение наследственной мышечной дистрофии Дюшенна.
статочно часто применяют (Преднизолон, Дефлазакорт), которые помогают замедлить патологический процесс в мышечных волокнах. Терапевтический курс стероидными препаратами способствует увеличению мышечной силы и уменьшению выраженности некоторых клинических симптомов. Однако эффект от их применения сохраняется непродолжительное время и высок риск возникновения побочных реакций.
Кроме того, были проведены клинические исследования по использованию лекарственных препаратов из группы бета-2-агонистов. У пациентов с миопатией Дюшенна они увеличивали мышечную силу, но не замедляли прогрессирование болезни. Динамический контроль проводился в течение года. Поэтому трудно говорить о долгосрочном эффекте применения этой группы препаратов для лечения наследственной патологии.
Поддержка дыхания

Прогрессирование заболевания неизбежно приводит к появлению серьёзных проблем с дыханием, также как и при . Необходимость использования искусственной вентиляции лёгких определяют по уровню насыщенности крови кислородом. В настоящее время представлен широкий выбор различных портативных аппаратов, позволяющих сделать это в домашних условиях. Как правило, искусственная вентиляция лёгких уже требуется в подростковом возрасте. Но бывают случаи, когда и в 20 лет, пациенты не нуждаются в поддержке дыхательной функции.
Если дыхательная маска не обеспечивает достаточного насыщения крови кислородом, может быть проведена:
- Интубация (введение специальной трубки в трахею через нос или рот).
- Операция трахеостомия (введение трубки через разрез трахеи на передней поверхности шеи).
Продолжительность применения искусственной вентиляции лёгких зависит от функционирования дыхательной системы. При падении жизненной ёмкости лёгких ниже 30% от нормальных показателей следует постоянно пользоваться подобными устройствами. Современные виды транспортных аппаратов искусственной вентиляции достаточно компактны и удобны в эксплуатации.
По уровню креатинфосфокиназы в крови можно судить о степени развития и прогрессирования мышечной дистрофии Дюшенна.
Лечение стволовыми клетками
Сегодня активно ведутся клинические исследования по разработке эффективного лечения от наследственной миопатии. Одно из перспективных направлений считается применение стволовых клеток. Учёные полагают, что эти клетки при определённых условиях способны будут заменить повреждённые мышечные волокна.
Кроме того, не менее перспективным является генная терапия. Например, немалый интерес для лечения наследственной мышечной дистрофии Дюшенна представляет активация гена, отвечающего за выработку утрофина. Как выяснилось, этот белок, по сути, считается аналогом дистрофина. Активировав продукцию утрофина, можно будет частично восполнить недостаток дистрофина в мышечных волокнах.
Лечебная физкультура

Каждому пациенту с миопатией Дюшенна показана лечебная физкультура, целью которой является предупреждение и замедление развития контрактур (ограничения подвижности в суставах), а также улучшение мышечного тонуса и силы. Начинать заниматься ЛФК необходимо как можно раньше, сразу после появления первых признаков патологии. Уровень физической нагрузки и комплекс упражнений определяют индивидуально, учитывая степень тяжести заболевания и общего состояния пациента.
Существуют отдельные реабилитационные центры, где целенаправленно занимаются с людьми, имеющими подобного рода нарушения. В среднем за год проходят 3–4 курса ЛФК. В перерывах между плановыми физиотерапевтическими курсами рекомендуют самостоятельные занятия лечебной физкультурой в домашних условиях. Большинство родителей после предварительного инструктажа со специалистом вполне справляются с этой задачей.
Если позволяет состояние пациента и имеется возможность, можно посещать бассейн. Плавание и упражнения в воде оказывают весьма благотворное влияние на организм ребёнка, страдающего столь тяжёлым недугом. Многие специалисты считают, что при отсутствии противопоказаний занятия в бассейне необходимо рекомендовать каждому пациенту с наследственной мышечной дистрофией.
Отсутствие умеренной физической активности способствует прогрессированию миопатии Дюшенна.
Массаж
В лечении мышечной дистрофии задействуют особые методики массажа. Добиться улучшения тонуса мышц является основной задачей массажиста. Рекомендуется систематически и регулярно проходить терапевтические курсы. В большинстве случаев врачи стараются обучить родственников стандартным методикам, чтобы параллельно можно было самостоятельно выполнять массаж в домашних условиях. Положительный эффект отмечается у пациентов, лечение которых включало сочетание занятий лечебной физкультурой, физиотерапевтических процедур и сеансов массажа.
Физиотерапия
Комплексное симптоматическое лечение миопатии Дюшенна практически всегда включает физиотерапевтические процедуры. На какой эффект можно рассчитывать от применения этих терапевтических методов:
- Активация метаболических процессов и улучшение трофики в мышечной ткани.
- Подавление дистрофических изменений в мышцах.
- Нормализация периферического кровообращения и микроциркуляции.
- Улучшение нервно-мышечной проводимости.
Пациентам с мышечной дистрофией могут назначать следующие физические методы лечения:
- Электрофорез.
- Лазеротерапия.
- Гидромассаж.
- Бальнеотерапия.
- Инфракрасное облучение.
- Ультрафонофорез.
Прогноз
При миопатии Дюшенна патологический процесс распространяется на все виды мышц: скелетные мышцы, миокард, гладкая мускулатура бронхов и др. Обычно средняя продолжительность жизни не превышает 30 лет. В единичных случаях пациенты с наследственной мышечной дистрофией могут дожить 40-летнего возраста. Правильная организация ухода за больным и использование всех современных средств, способных облегчить его состояние, позволяет увеличить продолжительность жизни.
Основным методом профилактики заболевания является пренатальная диагностика. Выявив серьёзную наследственную патологию на ранних сроках вынашивания плода, вы сможете сделать своевременное прерывание беременности.