Агаммаглобулинемия (Болезнь Брутона, Наследственная гипогаммаглобулинемия). Агаммаглобулинемия Брутона. Симптомы, диагностика, лечение Брутализм болезнь
– это наследственно обусловленное заболевание, при котором развивается тяжелый первичный иммунодефицит (дефект иммунной защиты организма) с выраженным снижением уровня гамма-глобулинов в крови. Проявляется болезнь обычно в первые месяцы и годы жизни ребенка, когда начинают развиваться повторные бактериальные инфекции: отит, синусит, пневмонии, пиодермии, менингит, сепсис. При обследовании в периферической крови и костном мозге практически отсутствуют сывороточные иммуноглобулины и B-клетки. Лечение агаммаглобулинемии заключается в пожизненной заместительной терапии.
МКБ-10
D80 Иммунодефициты с преимущественной недостаточностью антител

Общие сведения
Агаммаглобулинемия (наследственная гипогаммаглобулинемия, болезнь Брутона) – врожденный дефект гуморального иммунитета, обусловленный мутациями в геноме клеток, что приводит к недостаточности синтеза B-лимфоцитов. В результате нарушается образование иммуноглобулинов всех классов, и их содержание в крови резко снижается вплоть до полного отсутствия, развивается первичный иммунодефицит . Низкая реактивность иммунной системы приводит к развитию тяжелых повторных гнойно-воспалительных заболеваний ЛОР-органов, бронхов и легких, желудочно-кишечного тракта и мозговых оболочек. Болезнь Брутона встречается исключительно у мальчиков и наблюдается примерно у 1-5 человек из миллиона новорожденных, независимо от расы и этнической группы.
Причины
X-сцепленная форма наследственной агаммаглобулинемии возникает вследствие повреждения одного из генов X-хромосомы (расположен на Xq21.3-22.2). Этот ген ответственен за синтез фермента тирозинкиназы, участвующего в процессе образования и дифференцировки B-клеток. В результате мутаций этого гена и блокировки синтеза брутоновской тирозинкиназы нарушается формирование гуморального иммунитета. При агаммаглобулинемии молодые формы (пре-B-клетки) присутствуют в костном мозге, а их дальнейшая дифференцировка и поступление в кровеносное русло нарушена. Соответственно, выработка всех классов иммуноглобулинов практически не производится, и организм ребенка становится беззащитным при проникновении болезнетворных бактерий (чаще всего это стрептококки, стафилококки и синегнойная палочка).
Сходный механизм нарушений отмечается и в случае с другой формой наследственной агаммоглобулинемии - сцепленной с X-хромосомой и недостаточностью гормона роста. Аутосомно-рецессивная форма развивается в результате мутации нескольких генов (µ-тяжелых цепей, гена λ5/14.1, гена адапторного белка и гена сигнальной молекулы IgА).
Классификация
Выделяют три формы наследственной агаммаглобулинемии:
- сцепленная с X-хромосомой (85% всех случаев врожденных гипогаммаглобулинемий, болеют только мальчики)
- аутосомно-рецессивная спорадическая швейцарского типа (встречается у мальчиков и девочек)
- сцепленная с X-хромосомой и недостаточностью гормона роста агаммаглобулинемия (встречается крайне редко и только у мальчиков)
Симптомы агаммаглобулинемии
Сниженная реактивность гуморального иммунитета при агаммаглобулинемиях приводит к развитию повторных гнойно-воспалительных заболеваний уже на первом году жизни ребенка (как правило, после прекращения грудного вскармливания – в 6-8 месяцев). При этом защитные антитела от матери в организм ребенка уже не поступают, а свои иммуноглобулины – не вырабатываются. К 3-4 летнему возрасту воспалительные процессы переходят в хроническую форму со склонностью к генерализации. Гнойная инфекция при агаммаглобулинемии может поражать различные органы и системы.
Со стороны ЛОР-органов нередки гнойные гаймориты , этмоидиты , отиты, причем гнойный отит чаще развивается на первом году жизни ребенка, а синуситы – в 3-5 лет. Из заболеваний бронхолегочной системы наблюдаются повторные бронхиты , пневмонии , абсцессы легкого.
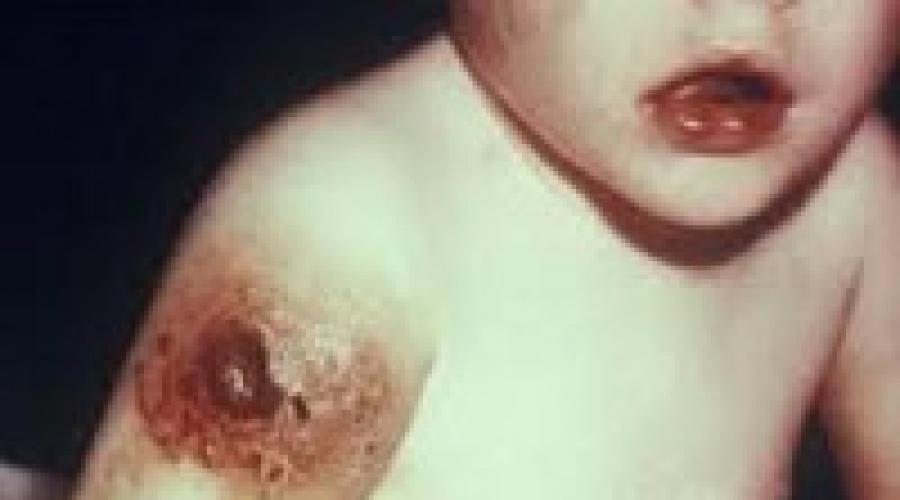
Часто встречается поражение желудочно-кишечного тракта с упорной диареей (поносами), вызванной хроническим инфекционным энтероколитом (основные возбудители – кампилобактерии, лямблии, ротавирус). На кожных покровах обнаруживается импетиго , микробная экзема , рецидивирующий фурункулез , абсцессы и флегмоны .
Нередким бывает поражение глаз (гнойные конъюнктивиты), полости рта (язвенные стоматиты , гингивиты), костно-мышечной системы (остеомиелиты , гнойные артриты). В целом клиническая картина агаммаглобулинемии характеризуется сочетанием общих симптомов, наблюдаемых при гнойной инфекции (высокая температура тела, озноб, боли в мышцах и головная боль, общая слабость, нарушение сна и аппетита и т. д.) и признаков поражения конкретного органа (кашель, одышка, затруднение носового дыхания, гнойное отделяемое, диарея и т. п.).
Осложнения
Любое инфекционное и соматическое заболевание у больного иммунодефицитом протекает тяжело, длительно и сопровождается осложнениями. Тяжелое течение агаммоглобулинемии может осложняться развитием менингита , вирусного энцефаломиелита, поствакцинального паралитического полиомиелита , сепсиса . На фоне заболевания повышена вероятность развития аутоиммунных и онкологических заболеваний. Гибель пациентов часто наступает от инфекционно-токсического шока.
Диагностика
При клиническом осмотре пациента врачом аллергологом-иммунологом выявляются признаки гнойно-воспалительного поражения того или иного органа (ткани) и симптомы, подтверждающие сниженную реактивность иммунной системы: гипоплазия миндалин, уменьшение периферических лимфатических узлов. Выражены и признаки отставания в физическом развитии ребенка.
Лабораторное исследование крови выявляет выраженное снижение уровня иммуноглобулинов в иммунограмме (IgA и IgM < 20 мг/дл – снижение в сто раз, IgG < 200 мг/дл – в десять раз) или их полное отсутствие. В периферической крови обнаруживается глубокий дефицит B-клеток (менее 1%), в костном мозге практически отсутствуют плазмоциты. Что касается клеточного иммунитета, то содержание T-лимфоцитов может быть в норме. При гистологическом исследовании лимфоидной ткани герминативные (зародышевые) центры и плазматические клетки отсутствуют.
Дифференциальная диагностика наследственной агаммаглобулинемии проводится с другими первичными и вторичными иммунодефицитными состояниями (генетическими нарушениями, ВИЧ и цитомегаловирусной инфекцией , врожденной краснухой и токсоплазмозом , злокачественными новообразованиями и системными нарушениями, иммунодефицитом вследствие интоксикации лекарственными препаратами и др.).
Лечение агаммаглобулинемии
Необходима пожизненная заместительная терапия антителосодержащими препаратами. Обычно используется введение внутривенного иммуноглобулина, а при его отсутствии – нативной плазмы от здоровых постоянных доноров. При впервые установленном диагнозе агаммаглобулинемии заместительное лечение проводится в режиме насыщения до достижения уровня иммуноглобулина IgG выше 400 мг/дл, после чего при отсутствии активного гнойно-воспалительного процесса в органах и тканях можно переходить к поддерживающей терапии с введением профилактических доз препаратов, содержащих иммуноглобулины.
Любой эпизод бактериальной гнойной инфекции, независимо от локализации воспалительного процесса, требует проведения адекватной антибактериальной терапии, которая выполняется одновременно с заместительным лечением. Чаще при агаммаглобулинемии используются антибактериальные средства из группы цефалоспоринов, аминогликозидов, макролидов, а также антибиотики пенициллинового ряда. Продолжительность лечения при этом в несколько раз превышает стандартную при данном заболевании.
Симптоматическое лечение проводится с учетом конкретного поражения того или иного органа (промывание околоносовых пазух носа антисептиками, выполнение вибрационного массажа грудной клетки и постурального дренажа при бронхитах и пневмониях и т. д.).
Прогноз
Если агаммаглобулинемия обнаружена в раннем возрасте до наступления тяжелых осложнений, и адекватная состоянию пациента заместительная терапия начата своевременно, возможно сохранение нормального образа жизни в течение многих лет. Однако в большинстве случаев диагностика наследственных нарушений гуморального иммунитета осуществляется слишком поздно, когда уже развились необратимые хронические гнойно-воспалительные заболевания органов и систем организма. В этом случае прогноз при агаммаглобулинемии неблагоприятный.
Болезнь Брутона (агаммаглобулинемия врожденная, Х-сцепленная агаммаглобулинемия) – первичный гуморальный иммунодефицит, который обусловлен мутационными изменениями в гене, кодирующем нерецепторную тирозинкиназу.
| МКБ-10 | D80.0 |
|---|---|
| МКБ-9 | 279.04 |
| DiseasesDB | 1728 |
| OMIM | 300300 |
| eMedicine | ped/294 derm/858 |
Общая информация
Х-сцепленная агаммаглобулинемия была впервые описана в 1952 году из США Огденом Брутоном. Доктор наблюдал за мальчиком, который с возраста 4 лет более десяти раз болел различными тяжелыми инфекционными патологиями, при этом в его крови не было обнаружено антител. Генетическая природа болезни Брутона была установлена в 1993 году.
Агаммаглобулинемии подвержены только представители мужского пола. Заболевание диагностируется у 1 мальчика из 250000.
Причины
Причина болезни Брутона – наличие мутантного белка в гене, кодирующем нерецепторную тирозинкиназу (тирозинкиназу Брутона или ТКБ). Этот ген расположен на одном из оснований Х-хромосомы, поэтому патологию называют Х-сцепленной.
Заболевание наследуется по рецессивному принципу. Проявляется оно только у мальчиков, так как в их ДНК присутствуют одна Х-хромосома и одна Y-хромосома. У девочек есть две Х-хромосомы, поэтому дефектный ген компенсируется нормальным. Но женщины являются носителями измененного гена и передают его сыновьям.
В редких случаях агаммаглобулинемия Брутона имеет ненаследственный характер: изменения в гене, кодирующем ТКБ, происходят после сразу зачатия.
Патогенез
Ген нерецепторной тирозинкиназы отвечает за созревание В-лимфоцитов – клеток, которые играют важную роль в работе гуморального иммунитета. При контакте с антигеном (вирусом или бактерией) одни из них превращаются в плазмоциты, которые продуцируют антитела (иммуноглобулины), а другие трансформируются в В-клетки памяти.
Брутоновская агаммаглобулинемия свидетельствует о поражении гуморального иммунитета. Из-за мутации гена ТКБ происходит блокировка процесса созревания В-лимфоцитов, в результате которой организм теряет способность полноценно вырабатывать иммуноглобулины при контакте с инфекционным агентом. Выраженность патологических изменений может варьироваться от существенного снижения уровня антител в крови до их полного отсутствия.
Как правило, у пациентов с Х-сцепленной агаммаглобулинемией в периферической крови обнаруживается незначительное или нулевое содержание В-лимфоцитов и низкая концентрация всех классов иммуноглобулинов, а также отсутствие плазматических клеток в лимфоидной ткани. Недостаточный синтез антител приводит к тому, что организм не справляется с инфекционными агентами, в частности с бактериями.
Для болезни Брутона характерно сохранение иммунного ответа на вирусные инфекции. Этим она отличается от генетической патологии под названием агаммаглобулинемия швейцарского типа, которой свойственен дефект клеточного и гуморального звеньев иммунитета. В основе ее развития лежит нехватка или отсутствие В- и Т-лимфоцитов, что делает человека подверженным инфекциям любой этиологии. Заболевание поражает представителей обоих полов.
Симптомы
Симптомы болезни Брутона начинают проявляться с 4-6 месяцев по мере того, как в крови ребенка снижается количество антител, переданных ему от матери. Основной признак агаммаглобулинемии с дефицитом В-клеток – хронические и рецидивирующие инфекции, спровоцированные пиогенными бактериями, то есть микроорганизмами, способными вызвать гнойное воспаление. К ним относятся пневмококки, стафилококки, гемофильные палочки и прочие.
Ребенок страдает от заболеваний ЛОР-органов, дыхательных путей, ЖКТ, кожи, подкожно-жировой клетчатки и так далее. Самые частые патологии – пневмония, отит, синусит, конъюнктивит, экзема, дерматомиозит, менингит, энцефалит.
Устойчивость к вирусам при болезни Брутона сохраняется, но ослабляется, из-за чего возникают осложнения. Вирус гепатита В приводит к прогрессирующему гепатиту, ротавирус – к синдрому мальабсорбции и хронической диарее, контакт с вирусом полиомиелита во время вакцинации – к полиомиелиту.
Дети с болезнью Брутона склонны к аллергическим реакциям, аутоиммунным заболеваниям, онкологическим патологиям, болезням соединительной ткани (артриту крупных суставов).
Другие проявления агаммаглобулинемии:
- отсутствие реакции со стороны лимфатической системы в острый период заболеваний;
- небольшой размер миндалин;
- бронхоэктазия – расширение бронхов, сопровождающееся приступами астмы.
У детей с дефектом гуморального иммунитета не бывает гиперплазии носоглоточной и небной миндалин.
Диагностика
Диагностируется болезнь Брутона на основании сбора анамнеза, осмотра пациента, инструментальных методов и лабораторных анализов. На патологию указывают частые бактериальные заболевания в анамнезе. Другие признаки агаммаглобулинемии, которые можно обнаружить с помощью рентгенографии: недоразвитие (отсутствие) миндалин и лимфоузлов, а также изменения в структуре селезенки.
Наиболее информативный метод диагностики болезни Брутона – исследование крови, в частности:
- проточная цитометрия – показывает пониженное количество или отсутствие В-лимфоцитов;
- сывороточный иммуноэлектрофорез – демонстрирует отсутствие фракции гамма-глобулинов;
- нефелометрия – позволяет выявить недостаточную концентрацию иммуноглобулинов (уровень Ig A и Ig M снижен в 100 раз, Ig G - в 10 раз);
- общий анализ – демонстрирует отклонение от нормы по числу лейкоцитов.
Кроме того, проводится молекулярно-генетическое исследование, во время которого обнаруживается дефект гена, кодирующего нерецепторную тирозинкиназу. Данный тест можно осуществить на этапе планирования или во время беременности.
Лечение
Основные принципы лечения агаммаглобулинемии – поддержание работы иммунной системы в течение всей жизни и антибиотикотерапия при развитии инфекционных патологий.
Для компенсации иммунодефицита проводится заместительная терапия препаратами гамма-глобулина. Лекарства вводятся внутривенно из расчета 400 мг на 1 кг массы тела. Цель фармакологического лечения – достижение концентрации иммуноглобулинов в крови равной 3 г/л.
В острый период инфекционных заболеваний при агаммаглобулинемии Брутона используются антибиотики: цефалоспорины, сульфаниламиды, пенициллины, аминогликозиды.
Прогноз
Болезнь Брутона имеет благоприятный прогноз при условии постоянного введения иммуноглобулинов и осуществления адекватной терапии антибиотиками. Несвоевременное применение антибактериальных средств при обострении инфекционных недугов может привести к стремительному прогрессированию патологического процесса и смертельному исходу.
Профилактика
Поскольку агаммаглобулинемия Брутона имеет генетическую природу, ее профилактика невозможна. Целесообразным является проведение генетического консультирования при планировании беременности парами, в семейном анамнезе которых есть данное заболевание.
Если у ребенка обнаружена агаммаглобулинемия, следует соблюдать меры по недопущению осложнений и рецидивов инфекций. К ним относятся:
- санация хронических очагов воспаления;
- адекватная терапия заболеваний;
- вакцинация только инактивированными препаратами.
Версия для печати
Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
Размещено на http://www.allbest.ru/
Реферат на тему:
«Болезнь и синдром Брутона»
ВВЕДЕНИЕ
1. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
2. ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ
4. БОЛЕЗНЬ БРУТОНА
4.1 Причины
4.2 Патофизиология
4.3 Симптомы и проявления
4.4 Диагностика
4.5 Лечение
4.6 Осложнения
4.7 Профилактика
ЗАКЛЮЧЕНИЕ
СПИСОК ЛИТЕРАТУРЫ
ВВЕДЕНИЕ
Иммунодефицитные состояния (ИДС) - стойкие или временные изменения иммунного статуса, обусловленные дефектом одного или нескольких механизмов иммунного ответа на антигенное воздействие.
1. ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Классификация иммунодефицитных состояний (ИДС)
I. По происхождению ИДС классифицируются следующим образом:
1) первичные (наследственные)
2) вторичные (приобретенные)
А) физиологические
Б) патологические
II. По механизмам развития выделяют следующие группы ИДС:
Первый механизм обусловлен
1) отсутствием или уменьшением числа вспомогательных клеток (АПК), т.е. мононуклеаров - макрофагов;
2) отсутствием или уменьшением числа лимфоцитов В-системы;
3) отсутствием или уменьшением числа T-лимфоцитов и их субпопуляций Т-системы;
4) отсутствием или уменьшением числа клеток все перечисленных категорий ИКС, т.е. комбинированные формы ИДС;
5) отсутствием или уменьшением клеток-предшественниц ИКС в связи с блокадой их созревания либо разрушения.
Второй механизм связывают с нарушениями процессов регуляции дифференцировки клеток В- и Т-систем, а также кооперации их и других клеток при реализации иммунного ответа.
III. По преимущественному повреждению клеток различных систем ИКС выделяют:
1)B-зависимые, или гуморальные ИДС;
2) T-зависимые, или клеточные ИДС;
3) фагоцитарные ИДС («А-зависимые»);
4) комбинированные ИДС - поражения клеточных и гуморальных механизмов иммунитета (например, В- и Т-лимфоцитов).
IV. Проявления нарушения иммунитета могут быть связаны:
с отсутствием, недостаточным количеством и/или ограничением функции клеток ИКС, а также нарушениями фагоцитоза и компонентов комплемента. Различные формы ИДС встречаются неодинаково - чаще всего повреждаются механизмы гуморального иммунитета, реже обнаруживаются нарушения клеточных и комбинированных формы иммунитета; остальные расстройства, получившие наименование неспецифических иммунодефицитных состояний (дефекты системы комплемента и фагоцитоза), встречаются крайне редко:
Гуморальные расстройства 75%
Комбинированные формы ИДС 10-25%
Дефекты клеточного иммунитета 5-10%
Нарушения функции фагоцитоза 1-2%
Дефект белков комплемента
2. ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ
Первичные иммунодефициты (наследуемые и врождённые дефекты иммунной системы) проявляются развитием инфекционных поражений организма вскоре после рождения, но могут не иметь клинических проявлений и до более позднего возраста.
Генные и хромосомные дефекты (многочисленные иммунодефициты разных классов).
Вторичные иммунодефициты, или иммунодефицитные состояния - иммунная недостаточность развивается вследствие эндо- и экзогенных воздействий на нормальную иммунную систему (например, около 90% всех вирусных инфекций сопровождается транзиторной иммунодепрессией).
Причины иммунодефицитных состояний многообразны, к ним отнесены:
Иммуносупрессивные препараты (включая фенитоин, пеницилламин, глюкокортикоиды).
Недостаточность питания, полостного и мембранного пищеварения, а также кишечного всасывания.
Наркотики и токсические вещества.
Лучевые воздействия, химиопрепараты.
Рост злокачественных опухолей.
Вирусы (например, ВИЧ).
Состояния, приводящие к потере белка (например, нефротический синдром).
Гипоксия.
Гипотиреоз.
Аспления.
Первичные иммунодефициты
В соответствии с номенклатурой ВОЗ под иммунологической недостаточностью первичного происхождения принято понимать генетически обусловленную неспособность организма реализовать то или иное звено иммунного ответа.
Согласно классификации, предложенной ВОЗ, в зависимости от преимущественного поражения В- и Т-звена иммунной системы, выделяют следующие первичные специфические иммунодефицитные состояния:
Комбинированные, с одновременным в одинаковой или разной степени выраженности повреждением клеточного (Т) и гуморального (В) звеньев иммунной системы;
С преимущественным повреждением клеточного (Т) звена иммунной системы;
С преимущественным повреждением гуморального (В) звена иммунной системы (патология продукции антител).
Первичные иммунодефицитные состояния встречаются, как правило, редко. По международной классификации болезней выделяют несколько разновидностей.
Тяжелый комбинированный Т- и В-иммунодефицитотличается развитием дефекта иммунокомпетентных структур на самых ранних этапах развития организма. Клинически протекает наиболее тяжело. Гибель организма может наступить внутриутробно, в первые дни после рождения из-за отсутствия или резкого угнетения стволовых и коммитированных гемопоэтических клеток, а также из-за отсутствия или резкою угнетения тимуса и других органов иммунной системы, с одновременным и выраженным снижением числа как Т-, так и В-лимфоцитов и плазматических клеток. Клинически проявляется резким снижением реактивности и резистентности организма к действию различных патогенных факторов, в том числе вирусов, бактерий, грибов.
Варианты комбинированных иммунодефицитных состояний обусловлены генетическими дефектами, затрагивающими различные линии дифференцировки лимфоцитов, а также ранние этапы их развития, общие для Т- и В-популяций.
Принципы лечения первичных ИДС
Лечение зависит от типа первичной иммунологический недостаточности и включает в себя целенаправленнуюзаместительную терапию (пересадка иммунокомпетентных тканей, трансплантация эмбрионального тимуса, костного мозга, введение готовых иммуноглобулинов - гглобулинов, концентрированных антител, прямое переливание крови от иммунизированных доноров, введение гормонов тимуса).
Применяется активная иммунизация против частых инфекций с помощью убитых вакцин, вводятся сульфаниламиды.
3. ВТОРИЧНЫЕ (ПРИОБРЕТЕННЫЕ) ИММУНОДЕФИЦИТЫ
Вторичные, или приобретенные, иммунодефициты - это нарушение иммунной защиты организма, встречающееся в постнатальном периоде в результате действия внешних или внутренних факторов, не связанное с первичным поражением генетического аппарата.
Вторичные иммунодефициты встречаются довольно часто.
Перечень основных заболеваний, сопровождающихся вторичным иммунодефицитом, предложенный экспертами ВОЗ.
1. Инфекционные заболевания:
а) протозойные и глистные болезни - малярия, токсоплазмоз, лейшманиоз, шистозоматоз и др.;
б) бактериальные инфекции - лепра, туберкулез, сифилис, пневмококковые, менингококковые инфекции;
в) вирусные инфекции - корь, краснуха, грипп, эпидемический паротит, ветряная оспа, острый и хронический гепатиты и др.;
г) грибковые инфекции - кандидоз, кокцидиодомикоз и др.
2. Нарушения питания - истощение, кахексия, нарушения кишечного всасывания и др.
3. Экзогенные и эндогенные интоксикации - при почечной и печеночной недостаточности, при отравлении гербицидами и др.
4. Опухоли лимфоретикулярной ткани (лимфолейкоз, тимома, лимфогрануломатоз), злокачественные новообразования любой локализации.
5. Болезни обмена (сахарный диабет и др.).
6. Потери белка при кишечных заболеваниях, при нефротическом синдроме, ожоговой болезни и др.
7. Действие различных видов излучения, особенно ионизирующей радиации.
8. Сильные, длительные стрессорные воздействия.
9. Действие лекарственных препаратов (иммунодепрессанты, кортикостероиды, антибиотики, сульфаниламиды, салицилаты и др.).
10. Блокада иммунными комплексами и антителами лимфоцитов при некоторыхаллергических и аутоиммунных заболеваниях.
Вторичные ИДС можно разделить на 2 основные формы:
1) системные, развивающиеся вследствие системного поражения иммуногенеза (при лучевых, токсических, инфекционных, стрессорных поражениях);
2) местные, характеризующиеся регионарным поражением иммунокомпетентных клеток (локальные нарушения иммунного аппарата слизистой, кожи и других тканей, развившиеся вследствие местных воспалительных, атрофических и гипоксических нарушений).
Физиологические иммунодефициты связаны с определенными причинами.
Недостаточность иммунной системы новорожденныхдетей характеризуется неполноценностью клеточного и гуморального звеньев, а также факторов неспецифической резистентности.
Большое количество лимфоцитов в периферической крови новорожденных сочетается со снижением функциональной активности Т- и В-лимфоцитов. Синтез антител на антиген идет преимущественно за счет IgM, содержание IgG и IgA снижено и достигает уровней взрослых людей только к 11-14-летнему возрасту. Отмечается низкая фагоцитарная активность и опсонизирующая способность крови. Уровень комплемента снижен и нормализуется к 3 --6-му месяцу жизни.
Иммунный статус беременных женщин отличается снижением числа и функций Т- и В-лимфоцитов, что можно объяснить увеличением содержания и активности Т-супрессоров. Это необходимо для подавления иммунного ответа на аллоантигены плода.
Недостаточность иммунной системы при старении проявляется в снижении активности как гуморального, так и клеточного звеньев. Снижаются уровни нормальных антител в крови, уменьшается способность к синтезу антител на антигенную стимуляцию. Это, в основном, низкоавидные антитела IgM; выработка антител IgG и IgA значительно уменьшена.
Угнетается синтез антител lgE, поэтому острота аллергических реакций смягчается. Существенно страдает клеточное звено иммунной системы. Снижается общее число лимфоцитов периферической крови, а также относительное и абсолютное число Т- и В-лимфоцитов и их функциональная активность. Уменьшается фагоцитарная активность макрофагов, нейтрофильных гранулоцитов, активность комплемента, лизоиима и бактерицидная активность сыворотки крови. Аутоиммунные реакции учащаются и с возрастом становятся более интенсивными.
Патологические иммунодефициты, также как и первичные (наследственные), характеризуются увеличением частоты (в десятки и даже сотни раз) злокачественных опухолей (ретикулосарком, лимфосарком и др.), особенно лейкозов, лимфом. При вторичных иммунодефицитах, как и при первичных, также может больше страдать либо гуморальный, либо клеточный иммунитет. Так, к развитию гуморального вторичного иммунодефицита часто приводят заболевания, сопровождающиеся потерей белков: ожоги, нефротический синдром, хронические нефриты и др.
К развитию клеточного вторичного иммунодефицита приводят тяжело протекающие вирусные инфекции (корь, грипп), грибковые заболевания (кандидозы наружные и внутренние).
Развитие иммунодефицита может происходить и по вине врачей, длительно использующих при трансплантации органов и лечении различных тяжелых заболеваний, особенно опухолевых, аутоиммунных (ревматоидного артрита), воспалительных иммунодепрессанты:
Кортикостероидные гормональные препараты;
Ингибиторы белкового синтеза;
Антибиотики;
Противоопухолевые цитостатики; антиметаболиты пуринового и пиримидинового ряда;
Рентгеновское облучение и др.
Принципы лечения вторичных ИДС
1. Заместительная терапия - использование различных иммунных препаратов (препаратов г-глобулина, антитоксических, антигриппозных, антистафилококковых сывороток и др.).
2. Коррекция эффекторного звена. Включает воздействие на иммунную систему фармакологическими препаратами, корригирующими ее работу (декарис, диуцефон, имуран, циклофосфамид и др.), гормонами и медиаторами иммунной системы (препараты тимуса - тимозин, тималин, Т-активин, лейкоцитарные интерфероны).
3. Выведение ингибирующих факторов, связывающих антитела и блокирующих эффект иммунокоррекции (гемосорбция,плазмаферез, гемодиализ, лимфоферез и др.).
Среди вторичных иммунодефицитов в последнее десятилетие приобретает все большее значение синдромприобретенногоиммунодефицита (СПИД). Последний был впервые описан в научной литературе в 1981 г. американскими учеными.
По данным ВОЗ, в последние годы число лиц с зарегистрированными случаями СПИДа удваивается за каждые полгода.
Если раньше число ВИЧ-инфицированных было в 50-- 100 раз больше, чем заболевших, то уже в 2001 г. число инфицированных достигло 130 млн человек, в том числе у 35 млн человек отмечались клинические проявления СПИДа.
4. БОЛЕЗНЬ БРУТОНА
Или агаммаглобулинемия Брутона, является наследственным иммунодефицитом, который вызывается мутациями в гене кодирующем тирозинкиназы Брутона. Впервые болезнь была описана Брутоном в 1952 году, в честь которого и был назван дефектный ген. Тирозинкиназы Брутона имеют решающее значение в созревании пре-В-клеток к дифференциации зрелых В-клеток. Ген тирозинкиназы Брутона был обнаружен на длинном плече Х-хромосомы в полосе от Xq21.3 к Xq22, он состоит из 37.5 килобаз с 19 экзонами, которые кодируют 659 аминокислот, именно эти аминокислоты завершают формирование цитозольной тирозинкиназы. В данном гене уже зафиксировано 341 уникальное молекулярное событие. В дополнение к мутациям, было обнаружено большое число вариантов или полиморфизмов.
иммунодефицитный синдром брутон инфекционный
4.1 Причины
Мутации в гене, лежащие в основе болезни Брутона, мешают развитию и функционированию В-лимфоцитов и их потомству. Основная идея заключается в том, что у здорового человека пре-В клетки созревают в лимфоциты. А у лиц страдающих этой болезнью, пре-В-клетки находятся или в малом количестве, или у них могут быть проблемы в функциональности.
4.2 Патофизиология
При отсутствии нормального белка, В-лимфоциты не дифференцируются или не созревают полностью. Без зрелых В-лимфоцитов, продуцирующие антитела плазмоциты также будут отсутствовать. Как следствие, ретикулоэндотелиальные и лимфоидные органы, в которых эти клетки пролиферируются,дифференцируются и хранятся, развиты плохо. Селезенка, миндалины, аденоиды, кишечник и периферические лимфоузлы, все они могут быть уменьшены в размере или вообще могут отсутствовать у лиц с Х-хромосомной агаммаглобулинемией.
Мутации в каждой из области гена могут привести к этой болезни. Самым распространенным генетическим событием является миссенс мутация. Большинство мутаций приводит к усечению белка. Эти мутации затрагивают критические остатки в цитоплазматическом белке и они весьма разнообразны и равномерно распределяются по всей молекуле. Тем не менее, тяжесть заболевания не может быть предсказана с помощью конкретных мутаций. Примерно одна треть точечных мутаций влияет на CGG сайты, которые, как правило содержат код для остатков аргинина.
Данный важный белок является необходимым для пролиферации и дифференцировки В-лимфоцитов. Мужчины с белковыми аномалиями имеют полное или почти полное отсутствие лимфоцитов в плазматических клетках.
4.3 Симптомы и проявления
Рецидивирующие инфекции начинают развиваться в раннем детстве и сохраняются на протяжении всей взрослой жизни.
Самым частым проявлением болезни Брутона или агаммаглобулинемии Брутона является увеличение восприимчивости к инкапсулированным гнойным бактериям, к таким как гемофильные инфекции, и некоторые виды Pseudomonas. Кожные инфекции у пациентов с болезнью в основном вызываются стрептококками группы А и стафилококками, они могут проявляться в виде импетиго, целлюлита, абсцессов, или фурункулов.
Форма экземы, которая напоминает атопический дерматит может быть очевидной, наряду с увеличением числа случаев гангренозной пиодермии, витилиго, алопеции и синдрома Стивенса-Джонсона (из-за более широкого использования препаратов). Другие инфекции, которые обычно присутствуют при этой болезни, включают энтеровирусные инфекции, сепсис, менингит и бактериальный понос. Пациенты также могут иметь аутоиммунные заболевания, тромбоцитопении, нейтропении, гемолитические анемии и ревматоидный артрит. Постоянныеэнтеровирусные инфекции очень редко приводят к смертельному энцефалиту или синдрому дерматомиозит-менингоэнцефалит. В дополнение к неврологическим изменениям, клинические проявления этого синдрома включают отеки и эритематозную сыпь на коже над разгибательными суставами.
Мужчины, могут развивать необычно тяжелый и / или периодический средний отит и пневмонии. Наиболее распространенным патогеном является S пневмония, а затем вирус гриппа B, стафилококки, менингококки и моракселла катаралис.
У детей в возрасте до 12 лет, типичные инфекции вызываются инкапсулированными бактериями. Общие инфекции в этой возрастной группе включают рецидивирующие пневмонии, синусит, и средний отит, которые вызываются S пневмония и вирусом гриппа В, которые трудно поддаются лечению в таком возрасте.
В зрелом возрасте, кожные проявления становятся более распространенными, как правило, из-за стафилококка и стрептококка группы А. Средний отит заменяется хроническим синуситом, и болезнь легких становится постоянной проблемой, как в ограничительной форме так и в обструктивной форме.
Как младенцы так и взрослые могут иметь аутоиммунные заболевания. Как правило, эти расстройства включают артрит, аутоиммунные гемолитические анемии, аутоиммунную тромбоцитопению, аутоиммунные нейтропении и воспалительные заболевания кишечника. Воспалительные заболевания кишечника могут быть очень трудно контролируемыми и они часто способствуют развитию хронической потери веса и недоеданию. Диарея является общей и вызывается Giardia или Campylobacter видами. Пациенты склонны к энтеровирусным инфекциям, в том числе к полиовирусу.
Физический осмотр
Младенцы мужского пола с агаммаглобулинемией Брутона, могут быть физически меньше, чем младенцы мужского пола без болезни из-за замедленного роста и развития от рецидивирующих инфекций.
При осмотре лимфатические узлы, миндалины, а также другие лимфоидные ткани могут быть очень маленькими или они вообще могут отсутствовать.
Заболевание диагностируется тогда, когда ребенку неоднократностановится плохо в присутствии различных инфекций, отита или стафилококковой инфекции кожи и конъюнктивита, которые не реагируют на терапию антибиотиками. Эти тяжелые инфекции могут быть связаны с нейтропенией.
Гангренозная пиодермия, например в виде язв и целлюлита нижних конечностей может также рассматриваться у некоторых пациентов.
4.4 Диагностика
Раннее выявление и диагностика имеет важное значение для предотвращения ранней заболеваемости и смерти от системных и легочных инфекций. Диагноз подтверждается аномально низкими уровнями или вообще отсутствующими зрелыми В-лимфоцитами, а также низкой или отсутствующей экспрессией тяжелой цепи м на поверхности лимфоцитов. С другой стороны, уровень Т-лимфоцитов будет повышенным. Окончательный определитель болезни - молекулярный анализ. Молекулярный анализ также используется для пренатальной диагностики, которая может быть выполнена с помощью отбора проб ворсинок хориона или амниоцентеза, когда мать, как известно, является носителем дефектного гена. Уровни IgG менее 100 мг/дл подтверждают диагноз.
Редко, но диагноз может быть поставлен у взрослых в их втором десятилетии жизни. Это, как полагают, происходит из-за мутации в белке, а не из-за его полного отсутствия.
Лабораторные анализы
На первом этапе необходимо провести количественное измерение IgG, IgM, иммуноглобулина Е (IgE) и иммуноглобулин А (IgA). Уровни IgG следует измерять во-первых, желательно после возраста 6 месяцев, когда уровни материнских IgG начнут снижаться. Во-вторых, уровни IgG ниже 100 мг/дл, как правило, свидетельствует о болезни Брутона. Как правило, IgM и IgA не обнаруживаются.
После того, как уровень антител будет определен как аномально низкий, подтверждение диагноза будет достигнуто с помощью анализа B-лимфоцитных и Т-лимфоцитных маркеров. Уровни CD19 + В-клеток ниже 100 мг/дл. Значения анализа Т-клеток (CD4 + и CD8 +), как правило, увеличиваются.
Дальнейший анализ может быть проведен путем обнаружения ответов IgG к Т-зависимым и Т-независимым антигенам,путем проведения иммунизации, например после введения неконъюгированной 23-валентной пневмококковой вакцины или вакцин от дифтерии, столбняка и H гриппа типа B.
Молекулярно-генетическим исследованием можно установить раннее подтверждение диагноза врожденной агаммаглобулинемии.
Другие тесты
Исследования функций легких занимают центральное место в мониторинге заболеваний легких. Они должны проводиться ежегодно у детей, которые могут выполнять тест (обычно от 5 лет).
Процедуры
Эндоскопия и колоноскопия могут быть использованы для оценки масштабов и прогрессирования воспалительного заболевания кишечника. Бронхоскопия может быть полезной в диагностике и отслеживании хронического заболевания легких и инфекций.
4.5 Лечение
Введение иммуноглобулина является основным методом контроля болезни. Типичные дозы - 400-600 мг/кг/мес должны назначаться каждые 3-4 недели. Дозы и интервалы могут быть скорректированы на основе отдельных клинических реакций. Терапия должна начинаться в возрасте 10-12 недель. Терапия с применением IgG должна начинаться с минимального уровня 500-800 мг/дл. Терапия должна начинаться в возрасте 10-12 недель.
Антибактериальная терапия Эпизоды бактериальных инфекционных осложнений при ВГГГ требуют антибактериальной терапии, как правило, парентеральной. Обязательным условием успеха антимикробной терапии при ВГГГ является ее одновременное применение с заместительной терапией, однако и в этом случае сроки антибактериальной терапии в 2-3 раза превосходят продолжительность стандартной антибиотикотерапии соответствующих воспалительных поражен органов в иммунокомпетентных пациентов. Дозировки антибиотиков остаются возрастными, но ориентированными на тяжелое и среднетяжелое течение инфекций.
Цефтриаксон может быть использован для лечения хронических инфекций, пневмонии, или сепсиса. Если это возможно, врачи должны получить культуры для выяснения чувствительности антибиотиков, так многие организмы уже сейчас проявляют устойчивость к многим антибиотикам. Инфекциистрептококка, в частности, могут потребовать цефтриаксона, цефотаксима или ванкомицина.
Бронхорасширители, стероидные ингаляторы, а также регулярные исследования функций легких (по крайней мере 3-4 раза в год) могут быть необходимой частью терапии в дополнение к антибиотикам.
Хронические дерматологические проявления атопического дерматита и экземы контролируются ежедневным увлажнением кожи специальными лосьонами и стероидами.
Хирургия
Хирургическое вмешательство может быть ограничено тяжелыми острыми инфекциями. Наиболее распространенные процедуры включают те, которые применяются для лечения пациентов с рецидивирующим отитом и с хроническим синуситом.
4.6 Осложнения
Осложнения включают хронические инфекции, энтеровирусные инфекции центральной нервной системы, увеличение частоты развития аутоиммунных заболеваний, а также инфекции кожи. Пациенты имеют повышенный риск развития лимфомы.
4.7 Профилактика
Поскольку агаммаглобулинемия Брутона имеет генетическую природу, ее профилактика невозможна. Целесообразным является проведение генетического консультирования при планировании беременности парами, в семейном анамнезе которых есть данное заболевание.
ЗАКЛЮЧЕНИЕ
Болезнь Брутона имеет благоприятный прогноз при условии постоянного введения иммуноглобулинов и осуществления адекватной терапии антибиотиками. Несвоевременное применение антибактериальных средств при обострении инфекционных недугов может привести к стремительному прогрессированию патологического процесса и смертельному исходу.
СПИСОК ЛИТЕРАТУРЫ
1. Аллергология и иммунология: национальное руководство / под ред. Р.М. Хаитова, Н.И. Ильиной. - М.: ГЭОТАР-медиа, 2009. - 656 с.
2. Педиатрия, Шабалов Н. П., СПб.: Спецлит, 2005.
3. Иммунология детского возраста: практическое руководство по детским болезням под ред. А. Ю. Щербины и Е. Д. Пашанова. М.: Медпрактика-М, 2006.
Размещено на Allbest.ru
Подобные документы
Болезни, вызванные недостаточностью иммунной системы. Болезни, обусловленные избыточным реагированием иммунной системы. Инфекции и опухоли иммунной системы. Классификация первичных иммунодефицитов по механизмам развития. Развитие болезни Брутона.
презентация , добавлен 19.04.2013
Вторичные иммунодефициты, клинические признаки, причины. Вирус иммунодефицита человека, пути передачи, стадии, лечение. Особенности течения ВИЧ-инфекции у детей. Аутовоспалительный синдром, классификация. Особенности диагностики врожденной ВИЧ-инфекции.
презентация , добавлен 15.03.2015
Характеристика системы иммунной защиты организма. Приобретенный иммунитет и его формы. Выработка антител и регуляция их продукции. Образование клеток иммунологической памяти. Возрастные особенности иммунитета, вторичные (приобретенные) иммунодефициты.
реферат , добавлен 11.04.2010
Иммунодефициты как нарушения иммунологической реактивности; классификация иммунодефицитных состояний. Причины и особенности вторичных ИДС: формы, инфекционные агенты, заболевания, лечение. Воздействие окружающей среды на возникновение вторичных ИДС.
презентация , добавлен 08.09.2014
Аутоаллергия: понятие и механизмы развития. Первичные и вторичные иммунодефициты. ВИЧ-инфекция: сущность, этиология, патогенез, механизм проявлений. Патогенетические принципы коррекции нарушений жизнедеятельности человеческого организма при СПИДЕ.
презентация , добавлен 11.11.2014
Сущность и этиология, признаки, классификация первичных иммунодефицитов. Локализация генетических дефектов при ПИ. Характерные инфекционные проявления. Вторичные иммунодефициты. Показания к применению некоторых препаратов. Профилактика и лечение инфекций.
презентация , добавлен 21.12.2014
Факторы риска наследственного заболевания. Синдром "кошачьего крика", его причины и симптомы. Синдром Лежена - врожденный комплекс пороков развития, обусловленный нарушением структуры одной из хромосом группы В. Профилактика наследственных заболеваний.
презентация , добавлен 09.04.2017
Первичные иммунодефициты: комбинированные, Т-клеточные, В-клеточные, дефекты системы мононуклеарных фагоцитов и гранулоцитов, недостаточность системы комплемента. Вторичные иммунодефициты: вирусные, при заболеваниях, при нарушении обмена веществ.
реферат , добавлен 18.08.2014
Клиническая картина железодефицитной анемии. Объективно анемический синдром. Прелатентный дефицит железа. Анемический синдром при мегалобластной анемии. Наследственный микросфероцитоз (болезнь Минковского-Шаффара). Нарушение синтеза спектрина и анкирина.
курс лекций , добавлен 03.07.2013
Синдром приобретенного иммунодефицита (СПИД). Система защиты организма от инфекций. Основные источники заражения и пути передачи инфекции. Опасность СПИД для организма человека. Применение антиретровирусной терапии. Самые распространенные мифы о СПИДе.